
.
Spinodal decomposition
Spinodal decomposition is a mechanism by which a solution of two or more components can separate into distinct regions (or phases) with distinctly different chemical compositions and physical properties. This mechanism differs from classical nucleation in that phase separation due to spinodal decomposition is much more subtle, and occurs uniformly throughout the material—not just at discrete nucleation sites.
Spinodal decomposition is of interest for two primary reasons. In the first place, it is one of the few phase transformations in solids for which there is any plausible quantitative theory. The reason for this is the inherent simplicity of the reaction. Since there is no thermodynamic barrier to the reaction inside of the spinodal region, the decomposition is determined solely by diffusion.[citation needed] Thus, it can be treated purely as a diffusional problem, and many of the characteristics of the decomposition can be described by an approximate analytical solution to the general diffusion equation.
In contrast, theories of nucleation and growth have to invoke the thermodynamics of fluctuations.[citation needed] And the diffusional problem involved in the growth of the nucleus is far more difficult to solve, because it is unrealistic to linearize the diffusion equation.
From a more practical standpoint, spinodal decomposition provides a means of producing a very finely dispersed microstructure that can significantly enhance the physical properties of the material.
Microstructural evolution under the Cahn–Hilliard equation, demonstrating distinctive coarsening and phase separation.
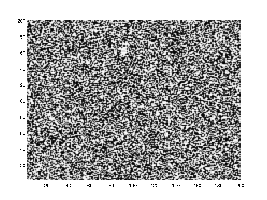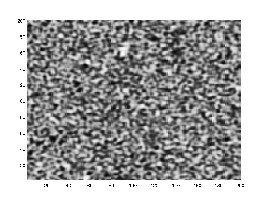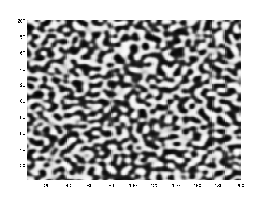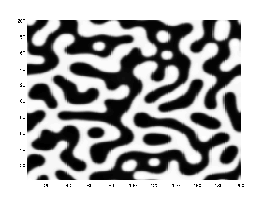
Microstructural evolution under the Cahn–Hilliard equation, demonstrating distinctive coarsening and phase separation.
Early evidence
In the early 1940s, Bradley reported the observation of sidebands around the Bragg peaks of the x-ray diffraction pattern from a Cu-Ni-Fe alloy that had been quenched and then annealed inside the miscibility gap. Further observations on the same alloy were made by Daniel and Lipson, who demonstrated that the sidebands could be explained by a periodic modulation of composition in the <100> directions. From the spacing of the sidebands they were able to determine the wavelength of the modulation, which was of the order of 100 angstroms.
The growth of a composition modulation in an initially homogeneous alloy implies uphill diffusion, or a negative diffusion coefficient. Becker and Dehlinger had already predicted a negative diffusivity inside the spinodal region of a binary system. But their treatments could not account for the growth of a modulation of particular wavelength, such as was observed in the Cu-Ni-Fe alloy. In fact, any model based on Fick's law yields a physically unacceptable solution when the diffusion coefficient is negative.
The first explanation of the periodicity was given by Mats Hillert in his 1955 Doctoral Dissertation at MIT. Starting with a regular solution model, he derived a flux equation for one-dimensional diffusion on a discrete lattice. This equation differed from the usual one by the inclusion of a term which allowed for the effect on the driving force of the interfacial energy between adjacent interatomic planes that differed in composition. Hillert solved the flux equation numerically and found that inside the spinodal it yielded a periodic variation of composition with distance. Furthermore, the wavelength of the modulation was of the same order as that observed in the Cu-Ni-Fe alloys. [1] [2]
A more flexible continuum model was subsequently developed by John W. Cahn, who included the effects of coherency strains as well as the gradient energy term. The strains are significant in that they dictate the ultimate morphology of the decomposition in anisotropic materials. [3] [4] [5]
Gibbs criteria
A metastable phase lies at a local but not global minimum in free energy, and is resistant to small fluctuations. J. Willard Gibbs described two criteria for a metastable phase: that it must remain stable against a small change over a large area, and that it must remain stable against a large change over a small area.[6]
Gradient energy
Gradient energies associated with even the smallest of compositional fluctuations can be evaluated using an approximation introduced by Ginzburg and Landau in order to describe magnetic field gradients in superconductors. This approach allows one to approximate the energy associated with a concentration gradient \nablaC. Thus, as a result of series expansions with respect to ( c – co ), this energy can be expressed in the form \( κ(\nablaC)^2 \) [7]
Note: In a three-dimensional Cartesian coordinate system R3 with coordinates ( x, y, z ), del is defined in terms of partial derivative operators as
\( \nabla = \mathbf{\hat{x}}{\partial \over \partial x} + \mathbf{\hat{y}}{\partial \over \partial y} + \mathbf{\hat{z}}{\partial \over \partial z} \)
\( \{\mathbf{\hat{x}}, \mathbf{\hat{y}},\mathbf{\hat{z}} \} \)are the unit vectors in the respective coordinate directions.
The vector derivative of a scalar field f is called the gradient, and it can be represented as:
\( \mbox{grad}\,c = {\partial c \over \partial x} \mathbf{\hat{x}} + {\partial c \over \partial y} \mathbf{\hat{y}} + {\partial c \over \partial z} \mathbf{\hat{z}} = \nabla c. \)
Cahn & Hilliard used such an approximation to evaluate the free energy of a small volume of non-uniform isotropic solid solution as follows:
\( dF = N_v [ F_0 + k( \nabla c)^2 ]~dV \)
or:
\( F = \int_v [ F_0 + k( \nabla c)^2 ]~dV \)
where:
\( N_v \) = particle density (#/vol)
\( F_0 \) is the free energy of the homogeneous solution.
The κ(\nablaC)2 term, is a measure of the free energy of a composition gradient and is strongly dependent on local composition. (The constant κ is related to derivatives of the free energy with respect to composition.) The interfacial energy associated with this compositional gradient therefore increases with the square of \nablaC. [8] [9]
Since we shall be concerned with testing the stability of an initially homogeneous solution to infinitesimal composition (or density) fluctuations, the gradients will also be infinitesimal and the second term will be completely sufficient to describe the contribution from the incipient 'surfaces" (between regions differing in composition). Higher order gradient energy terms will be negligible, except at very large gradients. We may also expand f (c) about the average composition co as follows:
\( f( c ) = f( c_o ) + \left( c - c_o \right) \left( \frac{\partial f}{\partial c} \right)_{c\,=\,c_o} + \frac12\, \left( c - c_o \right)^2 \left( \frac{\partial^2 f}{\partial c^2} \right)_{c\,=\,c_o}. \)
The difference in free energy per unit volume (or free energy density) between the initial homogeneous solution and one with a composition given by:
\( \left( c - c_o \right) = A\; \cos\, \beta x \, \)
is given by:
\( \frac{\Delta F}{V} = \left( \frac{A^2}{4} \right) \left[ \left( \frac{\partial^2 f}{\partial c^2} \right) + 2\, \kappa\, \beta^2 \right]. \)
Note that both terms are quadratic in the amplitude, so the stability criterion is initially independent of amplitude.
Thus, ΔF is positive if the second derivative of the free energy with respect to composition (hereafter referred to as f'' ) is positive, because the contribution of the surface energy in the second term is always positive. In this case, the system is stable against all infinitesimal fluctuations in composition since the formation of such fluctuations would result in an increase in the free energy of the system.
In contrast, if f'' is negative, then ΔF is negative when:
\( \left( c - c_o \right)^2\, \left( \frac{\partial^2 f}{\partial c^2} \right) > 2 \kappa \left(\nabla c\right)^2. \)
The formation of fluctuations can therefore be accompanied by a decrease in the free energy of the system within this region provided the scale or wavelength of the fluctuation is large enough. Within this context, such gradual changes in composition maintain small values for the gradient term \nablaC.
Fourier components
Cahn and Hilliard formulated a theory for the amplification (or attenuation) of an arbitrary composition fluctuation by considering, with Debye, the Fourier components of the composition rather than the composition itself. Thus, for a concentration fluctuation:
\( \left( c - c_o \right) = A\, \cos\, \beta x \)
one obtains for the change in free energy on forming fluctuations:
\( \frac{\Delta F}{V} = \left( \frac{A^2}{4} \right) \left(f'' + 2\, \kappa\, \beta^{2} \right). \)
The solution is then unstable (ΔF < 0) for all fluctuations of wave number β smaller than a critical wave number βc given by:
\( \beta_c = \sqrt{ \frac{f''}{2 \kappa} } \)
or for all fluctuations of wavelength λ = 2π/β which are longer than a critical wavelength given by:
\( \lambda_c = \sqrt{ \frac{8 \pi^2 \kappa}{f''} }. \)
From these equations, it is seen that the incipient surface energy, reflected in the gradient energy term, prevents the solution from decomposing on too small a scale. This concept was first introduced by Hillert, and shows that as the spinodal is approached, the critical wavelength approaches infinity. [8]
Phase diagram
This type of phase transformation is known as spinodal decomposition, and can be illustrated on a phase diagram exhibiting a miscibility gap. Thus, phase separation occurs whenever a material transitions into the unstable region of the phase diagram. The boundary of the unstable region, sometimes referred to as the binodal or coexistence curve, is found by performing a common tangent construction of the free-energy diagram. Inside the binodal is a region called the spinodal, which is found by determining where the curvature of the free-energy curve is negative. The binodal and spinodal meet at the critical point. It is when a material is moved into the spinodal region of the phase diagram that spinodal decomposition can occur. [10]
The free energy curve is plotted as a function of composition for a temperature below the convolute temperature, T". Equilibrium phase compositions are those corresponding to the free energy minima. Regions of negative curvature (∂2f/∂c2 < 0 ) lie within the inflection points of the curve (∂2f/∂c2 = 0 ) which are called the spinodes. Their locus as a function of temperature defines the spinodal curve. For compositions within the spinodal, a homogeneous solution is unstable against infinitesimal fluctuations in density or composition, and there is no thermodynamic barrier to the growth of a new phase. The spinodal therefore represents the limit of physical and chemical stability.
To reach the spinodal region of the phase diagram, a transition must take the material through the binodal region or the critical point. Often phase separation will occur via nucleation during this transition, and spinodal decomposition will not be observed. To observe spinodal decomposition, a very fast transition, often called a quench, is required to move from the stable to the spinodally unstable region of the phase diagram.
In some systems, ordering of the material leads to a compositional instability and this is known as a conditional spinodal, e.g. in the feldspars.[11][12][13][14] [15]
Diffusion equation
The mathematical theory of spinodal decomposition is based largely on the development of a generalized diffusion equation. [16] A diffusion equation relates a spontaneous flux of material to a gradient in composition. Fundamental thermodynamic principles dictate that in order for the flux to be spontaneous, it must be associated with a net decrease in the free energy of the system. Consider the following diffusion equation relating the flux of two species ( JA and JB ) to the gradient of the chemical potential difference:
\( - \tilde J = M \nabla (\mu_a - \mu_0) \)
As pointed out by Cahn, this equation can be considered as a phenomenological definition of the mobility M, which must by definition be positive. [17] It consists of the ratio of the flux to the local gradient in chemical potential.
The quantity ( μA - μB ) is the change in free energy when we reversibly add a unit amount of A atoms ( ΔF = + μA ) and simultaneously remove an equal number of B atoms ( ΔF = - μB ). This term may include factors such as composition, compositional gradients, stresses, and magnetic fields. For a homogeneous system:
\( \mu_a - \mu_b = \frac{\partial f}{\partial c} \)
The quantity f is the free energy of that number of lattice points in the crystal which initially occupied a unit volume. Substituting,
\( -J_a = M \frac{\partial^2 f}{\partial c^2} \nabla c \)
and defining the interdiffusion coefficient D by:
-\tilde J = - J_a = D \nabla_c \)
We can then define the interdiffusion coefficient D as follows:
\( D = M \frac{\partial^2 f}{\partial c^2} \)
Note that since M must always be positive, D takes its sign from the sign of f", which is negative within the spinodal. This has often been referred to as "uphill diffusion".
The above derivation of the diffusion coefficient is valid for concentration gradients that are so small that, for all practical purposes, each atom finds itself in surroundings which are similar to that which it would have in a homogeneous material of identical composition. If, however, concentration gradients are so large that within the range of interaction of an atom the average concentration has changed appreciably, then the atom will be aware of its inhomogeneous environment. This leads to a change in its chemical potential, and for fluids:
\( ( \mu_a - \mu_b ) =\frac{\partial f}{\partial c} - 2 K \nabla^2 c \)
Substitution yields:
\( - \tilde J = \mu \frac{\partial^2 f}{\partial c^2} \nabla c - 2 M K \nabla^3 c \)
By taking the divergence, we obtain the new diffusion equation:
\( \frac{\partial c} { \partial t} = M \frac{\partial^2 f}{\partial c^2} \nabla^2 c - 2 M k \nabla^4 c \)
Alternatively, since:
\( N_v (\mu_2 - \mu_1) = \frac {df}{dc} \)
the flux equation can be written as:
\( J = -M \left( \frac{d}{dx} \right) \frac{df}{dc} \)
For a system in equilibrium, the chemical potentials, and hence their difference, are constant throughout the system. Thus this equation for the flux satisfies the physical requirement that the net flux should go to zero as equilibrium is approached. For the time dependence of the composition we obtain on differentiation:
\( \frac{\partial c}{\partial t} = - \left( \frac{1}{N_v} \right) \left( \frac{\partial J}{\partial x} \right) = \left( \frac{m}{N_v} \right) f'' \frac{\partial^2 f}{\partial x^2} \)
Comparing this equation with the usual statement of Fick's second law
\( \frac{\partial c}{\partial t} = D \left( \frac{\partial^2 c}{\partial x^2} \right) \)
it is seen that the mobility is related to the interdiffusion coefficient by the following:
\( M = \frac{D N_v}{f''} \)
It then follows from the solution to be described next that a particular solution to this new diffusion equation is given by:
\( c -c_0 = A(\beta,t) \exp \left[i\beta x \right] \)
in which co is the average composition and A(β,t) is the amplitude of the Fourier component of wavenumber β at time t. In terms of the initial amplitude at time zero:
\( A(\beta , t ) = A(\beta , 0 ) \exp \left[ R(\beta)t \right] \)
where R(β) is an amplification factor given by:
\( R(\beta) = - \frac{M}{N} \beta^2 f'' \)
Coherency strains
For most crystalline solid solutions, there is a variation of lattice parameter with composition. If the lattice of such a solution is to remain coherent in the presence of a composition modulation, mechanical work has to be done in order to strain the rigid lattice structure. The maintenance of coherency thus affects the driving force for diffusion. [17] [18] [19] [20]
Consider a crystalline solid containing a one-dimensional composition modulation along the x-direction. We calculate the elastic strain energy for a cubic crystal by estimating the work required to deform a slice of material so that it can be added coherently to an existing slab of cross-sectional area. We will assume that the composition modulation is along the x' direction and, as indicated, a prime will be used to distinguish the reference axes from the standard axes of a cubic system (that is, along the <100>). [16]
Let the lattice spacing in the plane of the slab be ao and that of the undeformed slice a. If the slice is to be coherent after addition of the slab, it must be subjected to a strain δ in the z' and y' directions which is given by:
\( \epsilon = \frac{ a - a_0}{a_0} \)
In the first step, the slice is deformed hydrostatically in order to produce the required strains to the z' and y' directions. We use the linear compressibility of a cubic system 1 / ( c11 + 2 c12 ) where the c's are the elastic constants. The stresses required to produce a hydrostatic strain of δ are therefore given by:
\( \sigma_{x'} = \sigma_{y'} = \sigma_{z'} \)
The elastic work per unit volume is given by:
\( W_E = \frac{1}{2} \displaystyle \sum_i \sigma_i\epsilon_i \)
where the ε's are the strains. The work performed per unit volume of the slice during the first step is therefore given by:
\( W_E(1) = \frac{3}{2} ( c_{11} + 2 c_{12} ) \sigma^2 \)
In the second step, the sides of the slice parallel to the x' direction are clamped and the stress in this direction is relaxed reversibly. Thus, εz' = εy' = 0. The result is that:
\( W_E(2) = \frac{\sigma^2 (c_{11} + 2 c_{22})}{2c_{11}} \)
The net work performed on the slice in order to achieve coherency is given by:
\( W_E = W_E(1) - W_E(2) \)
or
\( W_E = \left( \frac{\sigma^2}{2} \right) (c_{11} + 2c_{12} ) \left( 3 - \left[ \frac{c_{11} - 2c_{12}}{c_{1'1'}} \right] \right) \)
The final step is to express c1'1' in terms of the constants referred to the standard axes. From the rotation of axes, we obtain the following:
\( c_{1'1'} = c_{11} + 2(2c_{44} - c_{11} + c_{12}) (l^2m^2 + m^2n^2 + l^2n^2) \)
where l, m, n are the direction cosines of the x' axis and, therefore the direction cosines of the composition modulation. Combining these, we obtain the following:
\( W_E = Y \sigma^2 \)
\( Y = \frac{1}{2} (c_{11} + 2c_{12}) \left[ 3 - \frac{c_{11} + 2c_{12}}{c_{11} + 2(2c_{44} - c_{11} + c_{12}})(l^2m^2 + m^2n^2 + l^2n^2) \right] \)
The existence of any shear strain has not been accounted for. Cahn considered this problem, and concluded that shear would be absent for modulations along <100>, <110>, <111> and that for other directions the effect of shear strains would be small. It then follows that the total elastic strain energy of a slab of cross-sectional area A is given by:
\( W_E = 4 \int Y \sigma^2~dx \)
We next have to relate the strain δ to the composition variation. Let ao be the lattice parameter of the unstrained solid of the average composition co. Using a Taylor's series expansion about co yields the following:
\( a = a_0[ 1 + \eta [c-c_0 ] + \cdots ] \)
in which
\( \eta = \left( \frac{1}{a_0} \right) \left(\frac{da}{dc}\right) + \frac{d \ln a}{dc} \)
where the derivatives are evaluated at co. Thus, neglecting higher order terms, we have:
\( \sigma = \frac{a-a_0}{a_0} = \eta ( c- c_0) \)
Substituting, we obtain:
\( W_E = A \int \eta^2 Y (c -c_0)^2~dx \)
This simple result indicates that the strain energy of a composition modulation depends only on the amplitude and is independent of the wavelength. For a given amplitude, the strain energy WE is proportional to Y. Let us consider a few special cases.
For an isotropic material:
\( 2c_{44} -c_{11} + c_{12} \)
so that:
\( Y[\mathrm{iso}] = c_{11} + c_{12} -2 (\frac{c_{12}^2}{c_{11}}) \)
Ths equation can also be written in terms of Young's modulus E and Poissons's ratio υ using the standard relationships:
\( c_{11} = \frac{ E (1-\nu)}{(1-2_\nu)(1 + \nu)} \)
\( c_{12} = \frac { E_\nu} {(1-2_\nu)(1+\nu)} \)
Substituting, we obtain the following:
\( Y[\mathrm{iso} ] = \frac{E}{1-\nu} \)
For most metals, the left hand side of this equation
\( 2c_{44} - c_{11} + c_{12} \)
is positive, so that the elastic energy will be a minimum for those directions that minimize the term: l2m2 + m2n2 + l2n2. By inspection, those are seen to be <100>. For this case:
\( Y[\mathrm{100}] = c_{11} + c_{12} -2 \left( \frac{c_{12}^2}{c_{11}}\right) \)
the same as for an isotropic material. At least one metal (molybdenum) has an anisotropy of opposite sign. In this case, the directions for minimum WE will be those that maximize the directional cosine function. These directions are <111>, and
\( Y[\mathrm{111}] = \frac{ 6c_{44} ( c_{11} + 2c_{12} )}{c_{11} + 2c_{12} + 4c_{44}} \)
As we will see, the growth rate of the modulations will be a maximum in the direcitons that minimize Y. These directions therefore determine the morphology and structural characteristics of the decomposition in cubic solid solutions.
Rewriting the diffusion equation and including the term derived for the elastic energy yields the following:
\( F_t = A \int f(c) + \eta Y (c-c_0)^2 + K\left(\frac{dc}{dx}\right)^2~dx \)
or
\( \frac{\partial c} {\partial t} = \left( \frac{M}{N_\nu}\right) \left( [ f'' + 2 \eta Y ] \left(\frac{d^2 c}{dx^2}\right) - 2K\left(\frac{d^4c}{dx^4}\right) \right) \)
which can alternatively be written in terms of the diffusion coefficient D as:
\( \frac{\partial c} {\partial t} = \left( \left[ 1 + \frac{ 2\eta Y}{f''} \right] \frac{d^2 c}{dx^2} - \frac{2KF}{f''} \frac{d^4c}{dx^4} \right) \)
The simplest way of solving this equation is by using the method of Fourier transforms.
Fourier transform
The motivation for the Fourier transform comes from the study of a Fourier series. In the study of a Fourier series, complicated periodic functions are written as the sum of simple waves mathematically represented by sines and cosines. Due to the properties of sine and cosine it is possible to recover the amount of each wave in the sum by an integral. In many cases it is desirable to use Euler's formula, which states that e2πiθ = cos 2πθ + i sin 2πθ, to write Fourier series in terms of the basic waves e2πiθ, with the distinct advantage of simplifying many unwieldy formulas.
The passage from sines and cosines to complex exponentials makes it necessary for the Fourier coefficients to be complex valued. The usual interpretation of this complex number is that it gives you both the amplitude (or size) of the wave present in the function and the phase (or the initial angle) of the wave. This passage also introduces the need for negative "frequencies". (E.G. If θ were measured in seconds then the waves e2πiθ and e−2πiθ would both complete one cycle per second—but they represent different frequencies in the Fourier transform. Hence, frequency no longer measures the number of cycles per unit time, but is closely related.)
If A(β) is the amplitude of a Fourier component of wavelength λ and wavenumber β = 2π/λ the spatial variation in composition can be expressed by the Fourier integral: [17]
\( c - c_0 = \int A(\beta) \exp (i \beta x)~d\beta \)
in which the coefficients are defined by the inverse relationship:
\( A(\beta) = \frac{1}{2\pi} \int (c-c_0) \exp(-i\beta x) ~dx \)
Substituting, we obtain on equating coefficients:
\( \frac{dA(\beta)}{dt} = - \frac{M}{N_\nu} [ f'' + 2 \eta^2Y + 2Y\beta^2 ] \beta^2 A(\beta) \)
This is an ordinary differential equation that has the solution:
\( A(\beta,t) = A(\beta,0) \exp[ R(\beta) t] \)
in which A(β) is the initial amplitude of the Fourier component of wave wavenumber β and R(β) defined by:
\( R(\beta) = - \frac{M}{N_\nu} (f '' + 2\eta Y + 2k\beta^2)\beta^2 \)
or, expressed in terms of the diffusion coefficient D:
\( R(\beta) = -\tilde{D} \left(1 + \frac{2\eta^2 Y}{f''} + \frac{2K}{f''}\beta^2 \right) \beta^2 \)
In a similar manner, the new diffusion equation:
\( \frac{\partial c }{ \partial t} = M \frac{\partial^2 f}{\partial c^2} \nabla^2 c - 2MK\nabla^4 c) \)
has a simple sine wave solution given by:
\( c - c_0 = exp[R\bar{\beta}t] cos\beta \cdot r \)
where R(β) is obtained by substituting this solution back into the diffusion equation as follows:
\( R(\bar{\beta}) - M\beta^2 \left( \frac{\partial^2 f}{\partial c^2} + 2 K \beta^2 \right) \)
For solids, the elastic strains resulting from (in)coherency add terms to the amplification factor R(β) as follows:
\( R(\bar{\beta}) = - M\beta^2 \left( \frac{\partial^2 f}{\partial c^2} + 2\eta^2 Y + 2K\beta^2 \right) \)
where, for isotropic solids:
\( Y = \frac{E}{1-\nu} \)
where E is Young's modulus of elasticity, υ is Poisson's ratio, and η is the linear strain per unit composition difference. For anisotropic solids, the elastic term depends on direction in a manner which can be predicted by elastic constants and how the lattice parameters vary with composition. For the cubic case, Y is a minimum for either (100) or (111) directions, depending only on the sign of the elastic anisotropy.
Thus, by describing any composition fluctuation in terms of its Fourier components, Cahn showed that a solution would be unstable with respect to sinusoidal fluctuations of a critical wavelength. By relating the elastic strain energy to the amplitudes of such fluctuations, he formalized the wavelength or frequency dependence of the growth of such fluctuations, and thus introduced the principle of selective amplification of Fourier components of certain wavelengths. The treatment yields the expected mean particle size or wavelength of the most rapidly growing fluctuation.
Thus, the amplitude of composition fluctuations should grow continuously until a metastable equilibrium is reached with a preferential amplification of components of particular wavelengths. The kinetic amplification factor R is negative when the solution is stable to the fluctuation, zero at the critical wavelength, and positive for longer wavelengths—exhibiting a maximum at exactly \( \sqrt{2} \) times the critical wavelength.
Consider a homogeneous solution within the spinodal. It will initially have a certain amount of fluctuation from the average composition which may be written as a Fourier integral. Each Fourier component of that fluctuation will grow or diminish according to its wavelength.
Because of the maximum in R as a function of wavelength, those components of the fluctuation with \(\sqrt{2} \) times the critical wavelength will grow fastest and will dominate. This "principle of selective amplification" depends on the initial presence of these wavelengths but does not critically depend on their exact amplitude relative to other wavelengths (if the time is large compared with (1/R). It does not depend on any additional assumptions, sinced different wavelengths can coexist and do not interfere with one another.
Limitations of this theory would appear to arise from this assumption and the absence of an expression formulated to account for irreversible processes during phase separation which may be associated with internal friction and entropy production. In practice, frictional damping is generally present and some of the energy is transformed into thermal energy. Thus, the amplitude and intensity of a 1-dimensional wave decreases with distance from the source, and for a three-dimensional wave the decrease will be greater.
Dynamics in k-space
In the spinodal region of the phase diagram, the free-energy can be lowered by allowing the components to separate, thus increasing the relative concentration of a component material in a particular region of the material. The concentration will continue to increase until the material reaches the stable part of the phase diagram. Very large regions of material will change their concentration slowly due to the amount of material which must be moved. Very small regions will shrink away due to the energy cost in maintaining an interface between two dissimilar component materials. [21] [22] [23]
To initiate a homogeneous quench a control parameter, such as temperature, is abruptly and globally changed. For a binary mixture of A-type and B-type materials, the Landau free-energy
\( F=\int\!\left(\frac{A}{2}\phi^2+\frac{B}{4}\phi^4 + \frac{\kappa}{2}\left(\nabla\phi\right)^2\right)~dx\;. \)
is a good approximation of the free-energy near the critical point and is often used to study homogeneous quenches. The mixture concentration \( \phi=\rho_A-\rho_B \) is the density difference of the mixture components, the control parameters which determine the stability of the mixture are A and B, and the interfacial energy cost is determined by \kappa.
Diffusive motion often dominates at the length-scale of spinodal decomposition. The equation of motion for a diffusive system is
\( \partial_t\phi=\nabla ( m\nabla\mu + \xi(x) )\;, \)
where m is the diffusive mobility, \( \xi(x) \) is some random noise such that \( \langle\xi(x)\rangle=0 \), and the chemical potential \mu is derived from the Landau free-energy:
\( \mu=\frac{\delta F}{\delta \phi}=A\phi+B\phi^3-\kappa \nabla^2 \phi\;. \)
We see that if A<0, small fluctuations around \phi=0 have a negative effective diffusive mobility and will grow rather than shrink. To understand the growth dynamics, we disregard the fluctuating currents due to \xi, linearize the equation of motion around \( \phi=\phi_{in} \) and perform a Fourier transform into k-space. This leads to
\( \partial_t\tilde{\phi}(k,t)=-m((A + 3B\phi_{in}^2)k^2 + \kappa k^4)\tilde{\phi}(k,t)=R(k)\tilde{\phi}(k,t)\;, \)
which has an exponential growth solution:
\( \tilde{\phi}(k,t) = \exp(R(k)t)\;. \) \)
Since the growth rate R(k) is exponential, the fastest growing angular wavenumber
\( k_{sp} = \sqrt{\frac{-(A+3B\phi_{in}^2)}{2\kappa}}\;, \)
will quickly dominate the morphology. We now see that spinodal decomposition results in domains of the characteristic length scale called the spinodal length:
\( \lambda_{sp} = \frac{2\pi}{k_{sp}} = 2\pi\sqrt{\frac{2\kappa}{-(A+3B\phi_{in}^2)}}\;. \)
The growth rate of the fastest growing angular wave number is
\( R(k_{sp})=-m((A + 3B\phi_{in}^2)k_{sp}^2 + \kappa k_{sp}^4)=\frac{m(A+3B\phi_{in}^2)^2}{4\kappa} = \frac{1}{t_{sp}} \)
where \( t_{sp} \) is known as the spinodal time.
The spinodal length and spinodal time can be used to nondimensionalize the equation of motion, resulting in universal scaling for spinodal decomposition.
References
^ Hillert, M., A Theory of Nucleation for Solid Metallic Solutions, Sc. D. Thesis (MIT, 1955)
^ Hillert, M., A Solid Solution Model for Inhomogeneous Systems, Acta Met., Vol. 9, p. 525 (1961)
^ Cahn, J.W., On spinodal decomposition, Acta Met., Vol. 9, p. 795 (1961)
^ Cahn, J.W., On spinodal decomposition in cubic crystals, Acta Met., Vol. 10, p. 179 (1962)
^ Cahn, J.W., Coherent fluctuations and nucleation in isotropic solids, Acta Met., Vol. 10, p. 907 (1962)
^ Gibbs, J.W., Scientific Papers of J Willard Gibbs, 2 vols. Bumstead, H. A., and Van Name, R. G., eds. (Dover, New York, 1961) ISBN 0918024773
^ Ginzburg, V.L. and Landau, L.D., J .Exptl. Theoret. Phys. (USSR), Vol. 20, p. 1064 (1950)
^ a b Cahn, J.W. and Hilliard, J.E., Free Energy of a Nonuniform System. I. Interfacial Free Energy, J. Chem. Phys., Vol. 28, p. 258 (1958)
^ Cahn, J.W. and Hilliard, J.E., Free Energy of a Nonuniform System. III. Nucleation in a Two-Component Incompressible Fluid, J. Chem. Phys., Vol. 31, p. 688 (1960)
^ Jones, Richard A. L. (2004) [2002]. Soft Condensed Matter. Oxford University Press. pp. 33. ISBN 0198505892. Retrieved 2007-10-22.
^ Cook, H.E., A lattice model of structural and dislocation transformations, Acta Met, Vol. 21, p. 1431 (1973)
^ Cook, H.E., On the nature of the omega transformation, Acta Met, Vol. 21, p. 1445 (1973)
^ Cook, H.E., On first-order structural phase transitions I. General considerations of pre-transition and nucleation phenomena, Acta Met., Vol. 23, p.1027 (1975)
^ Suzuki, T . and Wuttig, M., Analogy between spinodal decomposition and martensitic transformation, Acta Met., Vol. 23, p.1069 (1975)
^ Carpenter, M. A. (1981). "A "conditional spinodal" within the peristerite miscibility gap of plagioclase feldspars". Journal of the American Mineralogist 66: 553–560.
^ a b Hilliard, J.E., Spinodal Decomposition, in Phase Transformations p. 497 (American Society of Metals, Metals Park, 1970)
^ a b c Cahn, J.W., Spinodal Decomposition, 1967 Institute of Metals Lecture, Trans. Met. Soc. ASME, Vol. 242, p. 168 (1968)
^ De Fontaine, D., An approximate criterion for the loss of coherency in modulated structures, Acta Met., Vol. 17, p. 477 (1969)
^ Cook, H.E., De Fontaine, D., Hilliard, J.E., A model for diffusion on cubic lattices and its application to the early stages of ordering, Acta Met., Vol. 17, p. 765 (1969)
^ Cook, H.E. and De Fontaine, D., On the elastic free energy of solid solutions I. Microscopic theory, Acta Met., Vol. 17, p. 915 (1969)
^ De Fontaine, D., Mechanical instabilities in the b.c.c. lattice and the beta to omega phase transformation, Acta Met., Vol. 18, p. 275 (1970)
^ Cook, H.E. and De Fontaine, D., On the elastic free energy of solid solutions II. Influence of the effective modulus on precipitation from solution and the order-disorder reaction, Acta Met., Vol. 19, p. 607 (1971)
^ De Fontaine, D, Paton, N,E. and Williams, J.C., Acta Met., The omega phase transformation in titanium alloys as an example of displacement controlled reactions, Vol. 19, p. 1153 (1971)
Further reading
Langer, J.S.; Baron, M. and Miller, H.D. (1975). "New computational method in the theory of spinodal decomposition". Physical Review A 11 (4): 1417. Bibcode 1975PhRvA..11.1417L. doi:10.1103/PhysRevA.11.1417.
External links
Brief statement by Mats Hillert
John Cahn's Homepage
Binary alloys
Composition profiles
Copper / Nickel / Tin alloys
Graphical representation of microstructural evolution
Retrieved from "http://en.wikipedia.org/"
All text is available under the terms of the GNU Free Documentation License
