AGRICULTURAL AND BIOLOGICAL PUBLICATIONSCharles V. Piper, Consulting Editor THE CHEMISTRY
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

| Introduction | Page |
| Development of biological science; characteristics of protoplasm; plant and animal life, similarities and differences; protoplasmic activity essentially chemical changes; objects of study of the chemistry of plant life | xiii-xvi |
| CHAPTER I—Plant Nutrients | |
| Definitions; the plant food elements; available and unavailable forms; the value of the different soil elements as plant foods; functions of the different plant food elements in plant growth; inorganic plant toxins and stimulants; references | 1-15 |
| CHAPTER II—Organic Components of Plants | |
| Plants as synthetic agents; types of changes involved in plant growth; groups of organic compounds found in plants; physiological use and biological significance defined; physiological uses of organic groups | 16-20 |
| CHAPTER III—Photosynthesis | |
| Definitions; physiological steps in photosynthesis; formaldehyde, the simplest carbohydrate structure; the condensation of formaldehyde into sugars; theories concerning photosynthesis; the production of starches and sugars; references | 21-29 |
| CHAPTER IV—Carbohydrates | |
| Importance, nomenclature, and classification; groups of carbohydrates; isomeric forms of monosaccharides; chemical constitution of monosaccharides; characteristic reactions of hexoses; the occurrence and properties of monosaccharides; disaccharides; trisaccharides; tetrasaccharides; the relation of molecular configuration to biochemical properties; polysaccharides, dextrosans, levulosans, mannosans, and galactosans; physiological uses and biological significance of carbohydrates; references | 30-66 |
| CHAPTER V—Gums, Pectins, and Celluloses | |
| Relation to carbohydrates; groups; the natural gums and pentosans; mucilages; pectins; celluloses; physiological uses of celluloses; referencess | 67-75 |
| [Pg x]CHAPTER VI—Glucosides | |
| Definition; general structure; hydrolysis of the natural glucosides; general properties; the phenol glucosides; the alcohol glucosides; the aldehyde glucosides; the oxycumarin glucosides; the cyanophoric glucosides; the mustard-oil glucosides; the pigment glucosides; the digitalis glucosides; the saponins; physiological uses; biological significance; references | 76-93 |
| CHAPTER VII—Tannins | |
| General properties; occurrence; chemical constitution; classes; some common tannins; physiological uses; biological significance of tannins in fruits; references | 94-101 |
| CHAPTER VIII—Pigments | |
| Types and classes; the chlorophylls, chemical constitution, similarity of chlorophyll and hæmoglobin, properties of the chlorophylls; the carotinoids, carotin, xanthophyll, lycopersicin, and fucoxanthin; phycoerythrin and phycophæin; the anthocyans; the anthoxanthins; the production of ornamental pigments in flowers, etc.; the functions of pigments; references | 102-123 |
| CHAPTER IX—Organic Acids, Acid Salts, and Esters | |
| Chemical constitution; some common organic acids; physiological uses of organic acids; biological significance of fruit acids and esters | 124-128 |
| CHAPTER X—Fats and Oils, Waxes, and Lipoids | |
| General composition; fats and oils, occurrence, chemical constitution, acids which occur in natural fats, alcohols which occur in natural fats, hydrolysis and synthesis of fats, extraction of oils from plant tissues, identification of fats and oils, physiological use; the waxes; the lipoids, lecithin, other plant phosphatides, plant cerebrosides, physiological uses of lipoids; references | 129-145 |
| CHAPTER XI—Essential Oils and Resins | |
| Definitions, classes, occurrence; the essential oils; the resins; physiological uses and biological significance of essential oils; references | 146-150 |
| CHAPTER XII—The Vegetable Bases | |
| Composition and groups; the plant amines; alkaloids; the purine bases; the pyrimidines; the nucleic acids, composition and uses; references | 151-163 |
| CHAPTER XIII—Proteins | |
| Importance; general composition; amino-acids and peptid units; individual amino-acids; composition of the plant proteins; general properties of proteins; classification; differences between plant and animal proteins; extraction of proteins from plant tissues; synthesis in plants; physiological uses; references | 164-180 |
| [Pg xi]CHAPTER XIV—Enzymes | |
| Reaction velocities; enzymes as catalysts; general properties; extracellular and intracellular enzymes; chemical nature; nomenclature and classification; occurrence and preparation; general and individual enzymes; nature of enzyme action; accelerators and inhibitors; coenzymes and antienzymes; zymogens; physiological uses; further studies needed; references | 181-201 |
| CHAPTER XV—The Colloidal Condition | |
| "Colloids" and "crystalloids"; the colloidal condition a dispersion phenomenon; nomenclature and classification; conditions necessary to the formation of sols; gel-formation; general properties of colloidal solutions; suspensoids and emulsoids; adsorption; catalysis affected by the colloidal condition; industrial applications of colloidal phenomena; natural colloidal phenomena; references | 202-220 |
| CHAPTER XVI—The Physical Chemistry of Protoplasm | |
| Heterogeneous structure of protoplasm; protoplasm a colloidal gel; water; salts; osmotic pressure; surface boundary phenomena; electrical phenomena; acidity and alkalinity; summary; vital phenomena as chemical and physical changes; references | 221-238 |
| CHAPTER XVII—Hormones, Auximones, Vitamines, and Toxins | |
| External and internal stimulants; hormones; vitamines; auximones, toxins | 221-238 |
| CHAPTER XVIII—Adaptations | |
| General discussion; adaptations, accommodations, and adjustments; chromatic adaptations; morphological adaptations; accommodations; concluding statements | 249-258 |
| Index | 259-268 |
INTRODUCTION
The history of biological science shows that the conceptions which men have held concerning the nature of plant and animal growth have undergone a series of revolutionary changes as the technique of, and facilities for, scientific study have developed and improved. For a long time, it was thought that life processes were essentially different in character than those which take place in inanimate matter, and that the physical sciences had nothing to do with living changes. Then, too, earlier students had only vague notions of the actual structure of a living organism. Beginning with the earliest idea that a plant or an animal exists as a unit organism, to be studied as such, biological science progressed, first to the recognition and study of the individual organs which are contained within the organism; then to the tissues which make up these organs; then (with the coming into use of the microscope as an aid to these investigations) to the cells of which the tissues are composed; then to the protoplasm which constitutes the cell contents; and finally to the doctrine of organic evolution as the explanation of the genealogy of plants and animals, and the study of the relation of the principles of the physical sciences to the evolutionary process. The ultimate material into which organisms are resolved by this process of biological analysis is the cell protoplasm. But protoplasm is itself made up of a complex system of definite chemical compounds, which react and interact according to the laws of physical science. Hence, any study of the chemistry of plant growth is essentially a study of the chemical and physical changes which take place in the cell protoplasm.
Protoplasm differs from non-living matter in three respects. These are (1) its chemical composition; (2) its power of waste and repair and of growth; and (3) its reproductive power. From the standpoint of chemical composition, protoplasm is the most complex material in the universe. It not only contains a greater variety of chemical elements, united into molecules of enormous[Pg xiv] size and complexity, but also a greater variety of definite chemical compounds than exist in any other known mixture, either mineral or organic in type. One of the first problems in the study of protoplasm is, therefore, to bring this great variety of complex compounds into some orderly classification and to become familiar with their compositions and properties. Again, living matter is continually undergoing a process of breaking down as a result of its energetic activities and of simultaneously making good this loss by the manufacture of new protoplasm out of simple food materials. It also has the power of growth by the production of surplus protoplasm which fills new cells, which in turn produce new tissues and so increase the size and weight of individual organs and of the organism as a whole. Hence, a second field of study includes the chemical changes whereby new protoplasm and new tissue-building material are elaborated. Finally, living material not only repairs its own waste and produces new material of like character to it, but it also produces new masses of living matter, which when detached from the parent mass, eventually begin a separate existence and growth. Furthermore, the plant organism has acquired, by the process of evolution, the ability not only to produce an embryo for a successive generation but also to store up, in the tissues adjacent to it, reserve food material for the use of the young seedling until it shall have developed the ability to absorb and make use of its own external sources of food material. So that, finally, every study of plant chemistry must take into consideration the stored food material and the germinative process whereby this becomes available to the new organism of the next generation. Also, the chemistry of fertilization of the ovum, so that a new embryo will be produced, and the other stimuli which serve to induce the growth phenomena, must be brought under observation and study.
A further step in the development of biological science has been to separate the study of living things into the two sciences of botany and zoology. From the standpoint of the chemistry of the processes involved this segregation is unfortunate. It has resulted in the devotion of most of the study which has been given to life processes and living things to animal chemistry, or "physiological chemistry." As a consequence, biochemistry, which deals with the living processes of both plants and animals, is yet in its infancy; while phytochemistry is almost a new science,[Pg xv] yet its relation to the study of plants can scarcely be less vital than is that of physiological chemistry to studies of animal life.
The common conception that plant life and animal life are antithetical or complementary to each other has much to justify it. Animals breathe in oxygen and exhale carbon dioxide; while plants use the carbon dioxide of the air as a part of the raw material for photosynthesis and exhale oxygen. Plants absorb simple gases and mineral compounds as raw food materials and build these up into complex carbohydrates, proteins, fats, etc.; while animals use these complex compounds of plant origin as food, transforming parts of them into various other forms of structural material, but in the end breaking them down again into the simple gases and mineral compounds, which are expelled from the body through the excretory organs. Thus it would seem that the study of the chemistry of plant life and of animal life must necessarily deal with opposite types of phenomena.
But one cannot advance far into the study of the biochemistry of plants and animals before he discovers marked similarities in the chemical principles involved. Many of the compounds are identical in structure, undergo similar changes, and are acted upon by similar catalysts. Plant cells exhibit respiratory activities, using oxygen and giving off carbon dioxide, in exactly the same way that animal organisms do. The constructive photosynthetic processes of green plants are regulated and controlled by a pigment, chlorophyll, which is almost identical with the blood pigment, hæmatin", which regulates the vital activities in the animal organism, differing from the latter only in the mineral element which links the characteristic structural units together in the molecule. Many other points of similarity in the chemistry of the life processes of plants and animals will become apparent as the study progresses. It is sufficient now to call attention to the fact that these vital processes, in either plants or animals, are essentially chemical in character, and subject to study by the usual methods of biochemical investigations.
The protoplasm of the cell is the laboratory in which all the changes which constitute the vital activities of the plant take place. All of the processes which constitute these activities—assimilation, translocation, metabolism, and respiration—involve definite chemical changes. In so far as it is possible to study each of these activities independently of the others, they have been[Pg xiv] found to obey the ordinary laws of chemical reactions. Thus, the effect of the variations in intensity of light upon photosynthesis causes increase in the rate of this activity which may be represented by the ordinary responses of reaction velocities to external stimuli. Similarly, the effect of rises in temperature upon the rate of assimilation and upon respiration are precisely the same as their effect upon the velocity of any ordinary chemical reaction. Within certain definite ranges of temperature, the same statement holds true with reference to the rate of growth of the plant, although the range of temperature within which protoplasm lives and maintains its delicate adjustment to the four vital processes of life is limited; beyond a certain point, further rise in temperature does not produce more growth but rather throws the protoplasmic adjustment out of balance and growth either slows up markedly or stops altogether.
Hence, we may say that the methods by which the plant machine (protoplasm) accomplishes its results are essentially and definitely chemical in character and may be studied purely from the standpoint of chemical reactions, but the maintenance of the machine itself in proper working order is a vital phenomenon which is largely dependent upon the external environmental conditions under which the plant exists. A study of the phenomena resulting from the colloidal condition of matter is throwing a flood of light upon the mechanism by which protoplasm accomplishes its control of vital activities. But we are, as yet, a long way from a complete understanding of how colloidal protoplasm acquires and maintains its unique ability of self-regulation of the conditions necessary to preserve its colloidal properties and of how it elaborates the enzymes which control the velocity of the chemical reactions which take place within the protoplasm itself and which constitute the various processes of vital activity.
The object of this study of the chemistry of plant growth is to acquire a knowledge of the constitution of the compounds involved and of the conditions under which they will undergo the chemical changes which, taken all together, constitute the vital processes of cell protoplasm.
CHEMISTRY OF PLANT LIFE
CHAPTER I
PLANT NUTRIENTS
There is some confusion in the use of the terms "nutrient," "plant food," etc., as applied to the nutrition and growth of plants. Strictly speaking, these terms ought probably to be limited in their application to the organized compounds within the plant which it uses as sources of energy and of metabolizable material for the development of new cells and organs during its growth. Botanists quite commonly use the terms in this way. But students of the problems involved in the relation of soil elements to the growth of plants, including such practical questions as are involved in the maintenance of soil productivity and the use of commercial fertilizers for the growing of economic plants, or crops, are accustomed to use the terms "plant foods," or "mineral nutrients," to designate the chemical elements and simple gaseous compounds which are supplied to the plant as the raw material from which its food and tissue-building materials are synthetized. Common usage limits these terms to the soil elements; but there is no logical reason for segregating the raw materials derived from the soil from those derived from the atmosphere.
The essential difference between these raw materials for plant syntheses and the organic compounds which are produced within the plants and used by them, and by animals, as food, is that the former are inorganic and can furnish only materials but no energy to the organism; while the latter are organic and supply both materials and potential energy. It would probably be the best practice to confine the use of the word "food" to materials of the latter type, and several attempts have been made to limit its use[Pg 2] in this way and to apply some such term as "intake" to the simple raw materials which are taken into the organism and utilized by it in its synthetic processes. But the custom of using the words "food," or "nutrient," to represent anything that is taken into the organism and in any way utilized by it for its nourishment has been followed so long and the newer terms are themselves so subject to criticism that they have not yet generally supplanted the loosely used word "food."
If such use is permitted, however, it is necessary to recognize that only the green parts of green plants can use this inorganic "food," and that the colorless plants must have organic food.
To avoid this confusion, the suggestion has recently been made that all of the intake of plants and animals shall be considered as food, but that those forms which supply both materials and potential energy to the organism shall be designated as synergic foods, while those which contain no potential energy shall be known as anergic foods. On this basis, practically all of the food of animals, excepting the mineral salts and water, and all of the organic compounds which are synthetized by plants and later used by them for further metabolic changes, are synergic foods; while practically all of the intake of green plants is anergic food.
It is with the latter type of food materials that this chapter is to deal; while the following and all subsequent chapters deal with the organic compounds which are synthetized by plants and contain potential energy and are, therefore, capable of use as synergic food by either the plants themselves or by animals. It will be understood, therefore, that in this chapter the word "food" is used to mean the anergic food materials which are taken into and used by green plants as the raw materials for the synthesis of organic compounds, with the aid of solar energy, or that of previously produced synergic foods. In all later chapters, the term "food" will be used to mean the organic compounds which serve as the synergic food for the green parts of green plants and as the sole supply of nutrient material for the colorless parts of green plants and for parasitic or saprophytic forms (see page 16).
PLANT FOOD ELEMENTS
The raw materials from which the food and tissue-building compounds of plants are synthetized include carbon dioxide,[Pg 3] oxygen, water, nitrogen, phosphorus, sulfur, potassium, calcium, magnesium, and iron. The two gases first mentioned are derived directly from the air, through the respiratory organs of the plant. Water is taken into the plant chiefly from the soil, through its fibrous roots. All the other elements in the list are taken from the soil, nitrogen being derived from decaying organic matter (the original source of the nitrogen is, however, the atmosphere, from which the initial supply of nitrogen is obtained by direct assimilation by certain bacteria and perhaps other low forms of plant life), and the remaining ones from the mineral compounds of the soil.
Carbon dioxide and oxygen, being derived from the air, are always available to the leaves and stems of growing plants in unlimited supply; but the supply available to a seed when germinating in the soil, or to the roots of a growing farm crop, may sometimes become inadequate, especially in soils of a very compact texture, or "water-logged" soils. In such cases, the deficiency of these gaseous food elements may become a limiting factor in plant growth.
Water is often a limiting factor in plant growth. Experiments which have been repeated many times and under widely varying conditions show that when water is supplied to a plant in varying amounts, by increasing the percentage of water in the soil in which the plant is growing by regular increments up to the saturation point, the growth of the plant, or yield of the crop, increases up to a certain point and then falls off because the excess of water reduces the supply of air which is available to the plant roots. Hence, abundance of water is, in general, a most essential factor in plant growth.
Under normal conditions of air and moisture supply, however, the plant food elements which may be considered to be the limiting factors in the nutrition and growth of plants are the chemical elements mentioned in the list above.
AVAILABLE AND UNAVAILABLE FORMS
The plant food materials which are taken from the soil by a growing plant must enter it by osmosis through the semi-permeable membranes which constitute the epidermis of the root-hairs, and circulate through the plant either carried in solution in the sap or by osmosis from cell to cell. Hence, they must be in water-soluble[Pg 4] form before they can be utilized by plants. Obviously, therefore, only those compounds of these elements in the soil which are soluble in the soil water are available as plant food. The greater proportion of the soil elements are present there in the form of compounds which are so slightly soluble in water as to be unavailable to plants. The processes by which these practically insoluble compounds become gradually changed into soluble forms are chiefly the "weathering" action of air and water (particularly if the latter contains carbonic acid) and the action of the organic acids resulting from decaying animal or vegetable matter or secreted by living plants.
THE VALUE OF THE SOIL ELEMENTS AS PLANT FOOD
Analyses of the tissues of plants show that they contain all of the elements that are to be found in the soil on which they grew. Any of these elements which are present in the soil in soluble form are carried into the plants with the soil water in which they are dissolved, whether they are needed by the plant for its nutrition or not. But in the case of those elements which are not taken out of the sap to be used by the plant cells in their activities, the total amount taken from the soil is much less than is that of the elements which are used in the synthetic processes of the plant. Hence, much larger proportions of some elements than of others are taken from the soil by plants. The proportions of the different elements which are used by plants as raw materials for the manufacture of the products needed for their growth varies with the different species; but a certain amount of each of the so-called "essential elements" (see below) is necessary to every plant, because each such element has a definite rôle which it performs in the plant's growth. A plant cannot grow to maturity unless a sufficient supply of each essential element comes to it from the soil.
From the standpoint of their relative value as raw materials for plant food, the elements which are present in the soil may be divided into three classes; namely, the non-essential, the essential and abundant, and the critical elements.
The first class includes silicon, aluminium, sodium, manganese, and certain other rarer elements which sometimes are found in soils of some special type, or unusual origin. These elements seem[Pg 5] to have no rôle to play in the nutrition of plants; although silicon is always present in plant ash and sodium salts are found in small quantities in all parts of practically all plants. Nearly all species of plants can be grown to full maturity in the entire absence of these elements from their culture medium. Occasional exceptions to this statement in the case of special types of plants are known, and are of interest in special studies of plant adaptations, but need not be considered here.
The second group includes iron, calcium, magnesium, and, generally, sulfur. All of these elements are essential for plant growth, but are usually present in the soil in ample quantities to insure a sufficient supply in available form for all plant needs. Recent investigations have shown, however, that there are many soils in which sulfur is present in such limited quantities that many agricultural crops, when grown on these soils, respond favorably to the application of sulfur-containing fertilizers. In such cases, sulfur is a "critical" element.
The "critical" elements are those which are essential to the growth of all plants and which are present in most soils in relatively small proportions and any one may, therefore, be the limiting factor in plant growth so far as plant food is concerned. These are nitrogen, phosphorus, potassium, and (possibly) sulfur.
RÔLE OF PLANT FOOD ELEMENTS IN PLANT GROWTH
The use which a plant makes of the elements which come to it from the soil has been studied with great persistency and care by many plant physiologists and chemists. Many of the reactions which take place in a plant cell are extremely complicated, and the relation of the different chemical elements to these is not easily ascertained. It is probable that the same element may play a somewhat different rôle in different species of plants, in different organs of the same plant, or at different stages of the plant's development. But the usual and most important offices of each element are now fairly well understood, and are briefly summarized in the following paragraphs. It should be understood that a thorough and detailed discussion of these matters, such as would be included in an advanced study of plant nutrition, would reveal other functions than those which are presented here and would require a more careful and more exact method of statement than[Pg 6] is suitable here. However, the general principles of the utilization of soil elements by plants for their nutrition and growth may be fairly well understood from the following statements.
Nitrogen is a constituent of all proteins (see Chapter XIII). Proteins are apparently the active chemical components of protoplasm. Since it is in the protoplasm of the green portions, usually foliage, of plants that the photosynthesis of carbohydrates and the synthesis of most, or all, of the other tissue-building materials and reserve food substances of the plant takes place, the importance of nitrogen as a plant food can hardly be over-emphasized. Nitrogen starvation produces marked changes in the growth of a plant. Leaves are stunted in growth and a marked yellowing of the entire foliage takes place; in fact, the whole plant takes on a stunted or starved appearance. Abundance of nitrogen, on the other hand, produces a rank growth of foliage of a deep rich color and a luxuriant development of tissue, and retards the ripening process. In the early stages of growth, the nitrogen is present most largely in the leaves; but when the seeds develop, rapid translocation of protein material into the seeds takes place, until finally a large proportion of the total supply is deposited in them.
Nitrates are the normal form of nitrogen in the soil which is available to plants. During germination and early growth, the young seedling uses amino-acids, etc., derived from the proteins stored in the seed, as its source of nitrogen; and experiments have shown that similar forms of soluble organic nitrogen compounds can be successfully fed to the seedling as an external food supply. Soluble ammonium salts can be utilized as sources of nitrogen by most plants during later periods of growth, particularly by the legumes. But for most, if not all, of the common farm crops whose possibilities in these respects have been studied, it has been found that a unit of nitrogen taken up as a nitrate is very much more effective in promoting growth, etc., than is the same unit of nitrogen in the form of ammonium salts.
While the proteins are finally stored up largely in the seeds, or other storage organs, they are actively at work during the growing period in the cells of the foliage parts of the plant. Hence, the popular statement that "nitrogen makes foliage" is a fairly accurate expression of its rôle. Inordinate production of straw in cereal crops and of leaves in root crops often results from liberal supplies of available nitrogen in the soil early in the growing sea[Pg 7]son. If the crops develop to normal maturity, this excessive foliage growth has no harmful results, as the surplus material which has been elaborated is properly translocated into the desired storage organs; but, unfortunately, the retarding effect of the surplus nitrogen supply upon the date of maturing of the crop is often associated with premature ripening of the plants from other causes, with the consequence that too large a proportion of the valuable food material is left in the refuse foliage material of the crop. Crops which are grown solely for their leaves, such as hay crops, lettuce, cabbage, etc., profit greatly by abundant supplies of available nitrogen; although when foliage growth is stimulated in this way the tissue is likely to be thin-walled and soft rather than firm and solid.
Phosphorus is likewise an extremely important element in plant nutrition. But phosphorus starvation produces no such striking visible effects upon the growth of the plant as does lack of nitrogen. Abundance of available phosphorus early in the plant's life greatly stimulates root growth, and later on it undoubtedly hastens the ripening process; hence, this element seems to act as the exact antithesis of nitrogen.
The rôle of phosphorus, or of phosphates, in the physiological processes of the cell seems to be difficult to discover. The element itself is a constituent of some protein complexes and of the lecithin-like bodies (see page 141) which are supposed by some investigators to play an important part in determining the rate of chemical changes which take place in the cell and the movement of materials into and out of it. It is an essential constituent of the nucleus, and a meager supply of phosphorus retards, or inhibits, mitotic cell-division. Photosynthesis of sugars and the condensing of these into starch or cellulose takes place in plants in the absence of available phosphorus; but the change of these insoluble carbohydrates back again into soluble and available sugar foods does not.
Phosphorus is taken from the soil by plants in the form of phosphates. Much study has been given to the problem of the proper supply of available soil phosphates for economic crop production. Any discussion of soil fertility and fertilization which did not devote large attention to the conditions under which phosphates become available as plant food would be wholly inadequate; but such a discussion would be out of place here.[Pg 8]
The final result of an ample supply of phosphates in hastening the ripening process and stimulating seed production, as contrasted with that of an over-supply of nitrogen, has led to the popular statement that "phosphates make seeds." This statement, while not strictly accurate, is a fairly good summary of the combined results of the rôle of phosphorus in the plant economy. Large amounts of phosphorus are stored in the seeds. The two facts that large amounts of these compounds are thus available to the young seedling and that relatively large proportions of phosphates are taken from the soil by the plant during its early stages of growth are undoubtedly connected with the need for rapid cell-division at these periods in the plant's life.
Potassium.—The popular expression that "potash makes sugars and starch" is a surprisingly accurate description of the rôle of this element in plant metabolism. Either the photosynthesis of starch, or the changes necessary to its translocation (it is not yet certain which) is so dependent upon the presence of potassium in the cell sap that the whole process stops at once if an insufficient supply is present. The production and storage of sugar, or starch, in such root crops as beets, potatoes, etc., diminishes in direct proportion with a decreasing supply of potassium as plant food. The grains of the cereal crops become shrunken as a result of potassium starvation; and are plump and well filled with starch in the endosperm when sufficient potassium is available for the crop's needs.
The general tone and vigor of growth of the plant is largely dependent upon an ample potassium supply; potash-hungry plants, like those which have been weakened by any other unfavorable conditions, have been found to be more susceptible to injury by disease, than those which are well nourished with this food element. But potassium-starvation does not produce any pathological condition of the cell contents; its absence simply prevents the possibility of the development of the necessary carbohydrates for vigorous growth.
There is no known difference in the availability, or effectiveness, of potassium from the different forms of compounds containing it which may be present in the soil. Apparently, the only essential is that the compound shall be soluble so that it can be absorbed into the plant through the root-hairs. Of course, the acid radical to which the basic potassium ion is attached may, in[Pg 9] itself, have some beneficial or deleterious influence which gives to the compound as a whole some important effect in one case, which might not follow in the case of another type of compound; but the relative efficiency as plant food of a given unit of potassium seems to be the same regardless of the nature of the compound in which it is present.
Calcium is an essential plant food element but its physiological use has not yet been definitely established. It seems to stimulate root-development, and certainly gives vigor and tone to the whole plant. It is commonly believed that calcium is in some way connected with the development of cell-wall material. It has been reported that the stems of grasses and cereal plants become stiffer in the presence of ample calcium, but this may be due to greater turgidity rather than to strengthened cell-walls. Calcium remains in the leaves or stem as the plant ripens, but it is not clear that this has anything to do with the stiffness or weakness of the stem, or straw, of the plant. Experiments with algæ have shown that in the absence of calcium salts mitotic cell division takes place, showing that the nucleus functions properly, but the formation of the new transverse cell-wall is retarded. This is the only direct evidence that has been reported that calcium has any connection with cell-wall formation.
Certain species of plants, notably many legumes, require such large amounts of calcium salts for their growth as to give to them the popular appellation of "lime-loving plants." Other plants, known as "calciphiles," while not actually showing abnormally large percentages of calcium in their ash, flourish best on soils rich in lime. On the other hand, certain other species, known as "calcifuges," will not grow on soils which are even moderately rich in lime; in what respect these differ in their vital processes from others which demand large amounts of calcium, or those which flourish on soils rich in lime, has not been determined, however.
The beneficial effect of alkaline calcium compounds in the soil, in correcting injurious acidity, in improving the texture of clay soils, and in promoting the proper conditions for bacterial growth, is well known; but this has no direct connection with the rôle of calcium as plant food. Furthermore, calcium salts in the soil have a powerful influence in overcoming the harmful, or toxic, effects of excessive amounts of soluble salts of magnesium, sodium,[Pg 10] or potassium, in the so-called "alkali soils" (i.e., those which contain excessive amounts of water-soluble salts). The probable explanation for this fact is pointed out in a later paragraph of this chapter (see page 14); but this property of calcium probably has no connection with its physiological uses as plant food.
Magnesium, like phosphorus, is finally stored up mostly in the seeds, not remaining in the leaves and stems, as do calcium and potassium. This fact, together with other evidence obtained from experiments in growing plants in culture solutions containing varying amounts of this element, has led certain investigators to the conclusion that the rôle of magnesium is to aid in the transport of phosphorus, particularly from older to more rapidly growing parts of the plant. More recent investigations have shown, however, that magnesium has other roles which are probably more specific and more important than this one. It is now known that magnesium is a definite constituent of the chlorophyll molecule serving, as will be shown (see Chapter VIII), as the means of linkage between its essential component organic groups. Because of this fact, magnesium-starvation produces etiolated plants, which cannot function normally. Further, magnesium seems to be necessary for the formation of fats, apparently standing in a similar relation to fat-formation to that of potassium to carbohydrate-formation. This view is supported by the observations that when algæ are grown in magnesium-free solutions they contain no fat globules and that oily seeds are richer in magnesium than are those which store up starch as their reserve food material. Observers of the second of these phenomena have failed to note, however, that oily seeds are likewise richer in phosphorus than are starchy ones, and that the presence of larger proportions of magnesium in such seeds may, perhaps, be related to phosphorus-translocation rather than to fat-formation.
Whatever relation magnesium may have to fat-formation, or to the translocation of phosphorus, it is evident that these are rôles quite apart from its use as a constituent element in chlorophyll. As yet, no explanation of how it aids in these other synthetic processes has been advanced.
On the other hand, an excess of soluble magnesium salts in the soil produces definite toxic effects upon plants, magnesium compounds being known to be among the most destructive of the "alkali soil" salts. Calcium salts are remarkably efficient in[Pg 11] overcoming these harmful effects of magnesium salts. On this account, a large amount of experimental study has been given to the question of the calcium-magnesium ratio in plants. Numerous analyses of plant ashes have established the fact that there is a fairly definite ratio of this kind, which ratio, however, varies with the species of plant and is not correlated with the ratio of these elements present in the soil on which the plant grows, as was formerly believed. Cereal plants, as a rule, contain approximately twice as much lime as magnesia; while leafy plants (tobacco, cabbage, etc.) usually contain about four times as much calcium oxide as magnesium oxide.
Iron is essential to chlorophyll-formation. It is not a constituent of the chlorophyll molecule, as is magnesium; but in the absence of iron from the culture solution, a plant fails to produce chlorophyll and a green plant which is deprived of a supply of iron rapidly becomes etiolated. The way in which iron is related to chlorophyll-formation is not known.
Iron is taken from the soil by plants in the smallest proportions of any of the essential elements. Only soluble ferric compounds seem to serve as a suitable source of supply of the element; ferrous compounds being usually highly toxic to plants.
Sulfur is an essential element of plant food. The amounts required by plants were supposed, until recently, to be relatively small. This was due to the fact that earlier studies took account only of the sulfur which, on analysis, appeared as sulfates in the ash. Improved methods of analysis, which insure that the sulfur which is present in the plant tissue in organic combinations is oxidized under such conditions that it is not lost by volatilization during the combustion of the material, have shown that the total sulfur which is present in many plants approaches the quantity of phosphorus which is present in the same tissue. Furthermore, recent field and pot experiments have shown that at least a considerable part of the beneficial effects of many fertilizers, which has previously been attributed to the calcium, potassium, or phosphorus which they contain, is actually due to the sulfur present as sulfates in the fertilizers used.
Sulfur occurs in the organic compounds of plants, associated with phosphorus. It seems probable that its physiological uses are similar to those of the latter element; but there is as yet no experimental evidence to establish its exact rôle in the economy[Pg 12] of plant growth. It appears to be needed in largest proportion by plants which contain high percentages of nitrogen in their foliage, such as the legumes. There is some evidence that sulfur has a particular rôle in promoting the growth of bacteria, and it may be that the percentages of total sulfur which are found in the tissues of legumes are due to the presence of the symbiotic nitrogen-gathering bacteria in the nodules on the roots of these plants. This point has not yet been investigated, however.
Sodium is probably not essential to plant growth, although it is present in small proportions in the ash from practically all plants. In cases of insufficient supply of potassium, sodium can apparently perform at least a part of the rôle of the former element; but this seems not to be a normal relationship or use.
Chlorine is found in small amounts in the sap and in the ash of nearly all plants. However, it does not appear to be essential to the growth of a plant, except possibly in the case of certain species, such as asparagus, buckwheat, and, perhaps, turnips and some other root crops. Whether the benefit which these crops derive from the application of common salt to the soil in which they are growing is due to the direct food value of either the chlorine, or the sodium, or to some indirect effect, is not yet known. The presence of chlorine in the sap of plants is undoubtedly due to the inevitable absorption of soluble chlorides from the soil and apparently has no connection with the nutritional needs of the plant.
Silicon is always considered as a non-essential element, although it occurs in such large proportions in some plants as to indicate that it cannot be wholly useless. It accumulates in the stems of plants, chiefly in the cell-wall, and has sometimes been supposed to aid in giving stiffness to the stems. But large numbers of analyses have failed to show any direct correlation between the stiffness of straw of cereal plants and the percentage of silicon which they contain. Further, plants will grow to full maturity and with erect stems when no silicon is present in the mineral nutrients which are furnished to them. On the other hand, certain experiments appear to indicate that silicon can perform some of the functions of phosphorus, if soluble silicates are supplied to phosphorus-starved plants. But under normal conditions of plant nutrition, it seems to have no such function.[Pg 13]
INORGANIC PLANT TOXINS AND STIMULANTS
Much study has been given during recent years to the question of the supposed poisonous, or toxic, effects upon plants of various soil constituents. There seems to be no doubt that certain organic compounds which are injurious to plant life are often present in the soil, either as the normal excretions of plant roots or as products of the decomposition of preceding plant growths. A consideration of these supposedly toxic organic substances would be out of place in this discussion of mineral soil nutrients. But there seems to be no doubt that there may also be mineral substances in the soil which may sometimes exert deleterious influences upon plant growth. In fact, most metallic salts, except those of the few metals which are required for plant nutrition, appear to be toxic to plants. The exact nature of the physiological effects which are produced by these mineral toxins is not clearly understood; indeed, it is probably different in the case of different metals. Further, it is certain that both the stimulating and the toxic effect of metallic compounds upon low forms of plants is quite different from the effects of the same substances upon the more complex tissues of higher plants, a fact which is utilized to advantage in the application of fungicides for the control of parasitic growths on common farm crops.
Among the elements whose physiological effects upon higher plants, such as the cereal crops, etc., when their soluble compounds are present in the soil, have been carefully studied, there are three fairly distinct types of injurious mineral elements. The first of these, represented by copper, zinc, and arsenic, apparently exert their toxic effect regardless of the proportion in which they are present in the nutrient solution which is presented to the plant; although the degree of injury varies with the amount of injurious substance present, of course. The second type, of which boron and manganese are representatives, apparently exerts a definite stimulating effect upon plants when supplied to them in concentrations below certain clearly defined limits; but are toxic in concentrations above these. The third includes many soluble salts of magnesium, sodium, potassium, etc., which while either innocuous or else definite sources of essential plant foods when in lower concentrations, become highly toxic, or corrosive, when present in the soil solution in concentrations above the limits of "tolera[Pg 14]tion" of individual plants for these soluble salts. The tolerance shown by the different species of plants toward these soluble salts (the so-called "alkali" in soils) varies widely; indeed, there seems to be considerable variation in the resistance of different individual plants of the same species to injury from this cause.
With reference to the toxic effect of the third type of substances, i.e., the common soluble salts, it is known that single salts of potassium, magnesium, sodium, or calcium, in certain concentrations, are toxic to plants, while mixtures of the same salts in the same concentrations are not. Thus, solutions of sodium chloride, magnesium sulfate, potassium chloride, and calcium chloride which, when used singly, killed plants whose roots were immersed in them for only a few minutes, formed when mixed together a nutrient solution in which the same plants grew normally. The remarkable remedial effect of calcium salts in overcoming the injurious effects of other soluble salts has already been mentioned. One explanation of these relationships between mineral soil constituents and the living plant is that the life phenomena depend upon a balanced adjustment between the compounds of these different mineral elements with the proteins (producing the so-called "metal proteids") which constitute the active material of the cell protoplasm. According to this theory, any excess or deficiency of any one or more of these elements in the plant juices which surround a given cell will, of course, cause an interchange with the mineral components of the supposed "metal proteids" which upsets the assumed essential balance between them, with disastrous results. A more recent, and much more satisfactory, explanation of the "antagonism" between mineral elements in their toxic effects upon plants, which has both theoretical and experimental confirmation, is that single salts disturb the colloidal condition (see Chapter XV) of the protoplasm of the plant cells in such a way as to destroy its permeability to nutrient substances, while mixtures of salts restore the proper state of colloidal dispersion and permit the normal functioning of the protoplasm.
It is apparent from the above brief discussions that the rôle of the different soil elements as plant food, and their relations to the complex processes which constitute plant growth, afford an interesting and promising field for further study.[Pg 15]
References
Brenchley, Winifred E.—"Inorganic Plant Poisons and Stimulants," 106 pages, 18 figs., Cambridge, 1914.
Hall, A. D.—"Fertilizers and Manures," 384 pages, 7 plates, London, 1909.
Hall, A. D.—"The Book of the Rothamsted Experiments," 294 pages, 49 figs., 8 plates, London, 1905.
Hopkins, C. G.—"Soil Fertility and Permanent Agriculture," 653 pages, Chicago, 1910.
Hilgard, E. W.—"Soils," 593 pages, 89 figs., New York, 1906.
Loew, O.—"The Physiological Rôle of Mineral Nutrients," U. S. Department of Agriculture, Bureau of Plant Industry, Bulletin No. 45, 70 pages, Washington, D. C., 1903.
Russell, E. J.—"Soil Conditions and Plant Growth," 243 pages, 13 figs., Monographs on Biochemistry, London, 1917. (3d ed.)
Whitney, M.—"A Study of Crop Yields and Soil Composition in Relation to Soil Productivity," U. S. Department of Agriculture, Bureau of Soils, Bulletin No. 57, 127 pages, 24 figs., Washington, D. C., 1909.
CHAPTER II
THE ORGANIC COMPONENTS OF PLANTS
From the standpoint of their ability to synthetize synergic foods (see page 2) from inorganic raw materials, plants may be divided into two types; namely, the autotrophic, or self-nourishing, plants, and the heterotrophic plants.
Strictly speaking, only those plants whose every cell contains chlorophyll are entirely self-nourishing; and some parts, or organs, of almost any autotrophic plant are dependent upon the active green cells of other parts of the plant for their synergic food. Furthermore, if the term is used in a very wide sense, green plants are more than self-nourishing, they really nourish all living things. But the general significance of the term "autotrophic plants" is apparent.
"Heterotrophic plants" must, of necessity, get food, either directly or indirectly, from some other plant which can synthetize synergic foods or, in a few cases, from animal organic matter. If they do this by feeding upon the organic compounds of other living organisms, they are known as "parasites"; while if they secure their organic food from the tissues or debris of dead organisms, they are called "saprophytes." The heterotrophic plants are chiefly the bacteria and fungi; although a few seed-plants are devoid of chlorophyll or have nutritive habits similar to those of the non-green plants, and a few species are semi-parasitic or semi-saprophytic.
It is obvious that the metabolic processes of the autotrophic plants are very different from those of the heterotrophic type of plants. These differences constitute a most interesting field of study for plant physiologists. But the nature of the chemical compounds themselves and of the chemical changes involved in their transformations is not radically different in the two types of plants, the essential difference being in the preponderance of one kind of activities, or chemical reactions, over another in bringing about the metabolic processes which are characteristic of each[Pg 17] particular species. Hence, it does not seem necessary, or desirable, in this study of the chemistry of plant growth, to present as detailed a consideration of the differences in metabolic activity of the different types of plants as complete accuracy of statement in all cases might demand. We will, instead, discuss the organic chemical components of plant tissues and the reactions which they undergo, using the more common type of autotrophic plants as the illustrative material in most cases.
Hence, it will be understood that in all the following discussions of plant activities, except where specific exceptions are definitely mentioned, it is the green, or autotrophic, plants to which reference is made in each case.
From the standpoint of the sum total of its activities, a green plant is essentially an absorber of solar energy and a synthetizer of organic substances. Each individual autotrophic plant takes up certain amounts of the anergic foods which are discussed in the preceding chapter and manufactures from them a great variety of complex organic compounds, using the energy of the sun's rays, absorbed by chlorophyll, as the source for the energy necessary to accomplish these synthetic reactions. The ultimate object of these processes is to produce seeds, each containing an embryo and a sufficient supply of food for the young plant of the next generation to use until it has developed its own synthetic organs; or (in the case of perennials) to store up reserve food materials with which to start off new growth after a period of rest and often of defoliation. To be sure, animals and men often interfere with the completion of the life cycle of the plant, and utilize the seeds or stored food material for their own nutrition, but this is a biological relation which has no influence upon the nature of the plant's own activities.
Since all of these synthetic reactions must go on at ordinary temperatures, active catalyzers are necessary. These the plant provides in the form of enzymes (see Chapter XIV) which are always present in active plant protoplasm. Proper conditions for rapid chemical action are further assured by the colloidal nature (see Chapter XV) of the protoplasm itself.
TYPES OF CHEMICAL CHANGES INVOLVED IN PLANT GROWTH
The whole cycle of chemical changes which is involved in plant growth represents the net result of two opposite processes; the[Pg 18] first of these is a constructive one which has at least three different phases: namely, a synthesis of complex organic compounds, the translocation of this synthetized material to the centers of growth, and the building up of this food material into tissues or reserve supplies; and the second is a destructive process of respiration whereby carbohydrate material is broken down, potential energy is released, and carbon dioxide is excreted.
The synthetic processes which take place in plants are of two types; namely, photosynthesis, in which sugars are produced, and another, which has no specific name, whereby proteins are elaborated. The translocation of the synthetized material involves the change of insoluble compounds into soluble ones, effected by the aid of enzymes. For storage purposes, the soluble forms are usually, though not always, condensed again into more complex forms, these latter changes requiring much less energy than do the original syntheses from raw materials.
The destructive process, respiration, is characteristic of all living matter, either plant or animal organisms. It takes place continuously throughout the whole life of a plant. During rapid growth it is overshadowed by the results of the synthetic process, but during the ripening period in which the seed is matured, and during the germination of the seed itself, growth is practically at a standstill and the respiratory, destructive action predominates, so that the plant actually loses weight.
GROUPS OF ORGANIC COMPOUNDS FOUND IN PLANTS
As a result of their various synthetic and metabolic activities, a great variety of organic compounds is produced by plants. Certain types of these compounds, such as the carbohydrates and proteins, are necessary to all plants and are elaborated by all species of autotrophic plants. Other types of compounds are produced by many, but not all, species of plants; while still others are found in only a few species. It is fairly easy to classify all of these compounds into a few, well-defined groups, based upon similarity of chemical composition. These groups are known, respectively, as the carbohydrates and their derivatives, the glucosides and tannins; the fats and waxes; the essential oils and resins; organic acids and their salts; the proteins; the vegetable bases and alkaloids; and the pigments. A consideration of these[Pg 19] groups of compounds, as they are synthetized by plants, constitutes the major portion of the study of the chemistry of plant life as presented in this book. Following the discussion of the compounds themselves, the chapters dealing with enzymes, with the colloidal nature of protoplasm, and with the supposed accessory stimulating agencies, aim to show how the manufacturing machine known as the plant cell accomplishes its remarkable results, so far as the process is now understood.
PHYSIOLOGICAL USES AND BIOLOGICAL SIGNIFICANCE
In connection with the discussion of each of the above-mentioned groups of organic components of plants, an attempt will be made to point out what significance these particular compounds have in the plant's life and growth. Certain terms will be used to designate different rôles, which it is probably necessary to define.
There may be two possible explanations of, or reasons for, the presence of any given type of compound in the tissues of any particular species of plant. First, it may be supposed that this particular type of compounds is elaborated by the plant to satisfy its own physiological needs, or for the purpose of storing it up in the seeds as synergic food for the growth of the embryo, in order to reproduce the species. For this rôle of the various organic food materials, etc., we will employ the term "physiological use." On the other hand, it is often conceivable that certain types of compounds, which have properties that make them markedly attractive (or repellent) as a food for animals and men, or which are strongly antiseptic in character, or which have some other definite relationship to other living organisms, have had much to do with the survival of the particular species which elaborates them, in the competitive struggle for existence; or have been developed in the plant by the evolutionary process of "natural selection." For this relation of the compound to the plant's vital needs, we will use the term "biological significance." Such a segregation of the rôles which the different compounds play in the plant's economy may be more or less arbitrary in many cases; but it will be clear that when physiological uses are discussed, reference is being made to the plant's own internal needs; while the phrase biological significance will be understood to refer to the relation of the plant to other living organisms.[Pg 20]
PHYSIOLOGICAL USES OF THE ORGANIC COMPONENT GROUPS
From the standpoint of the rôle which each plays in the plant economy, the several groups of organic compounds may be roughly divided into three classes. These are: (a) the framework materials, including gums, pectins, and celluloses; (b) synergic foods, including carbohydrates, fats, and proteins; and (c) the secretions, including the glucosides, volatile oils, alkaloids, pigments, and enzymes.
The framework material, as the name indicates, constitutes the cell-wall and other skeleton substances of the plant. It is made up of carbohydrate complexes, produced by the cell protoplasm from the simpler carbohydrates.
The synergic foods, or "reserve foods" as they are sometimes called, produced by the excess of synthetized material over that needed for the immediate use of the plant, are accumulated either in the various storage organs, to be available for future use by the plant itself or by its vegetative offspring, or in the seed, to be available to the young seedling of the next generation. Proteins not only serve as reserve food materials but also make up the body of the living organism itself. Carbohydrates and fats serve as synergic and reserve foods.
The secretions may be produced either in ordinary cells and found in their vacuoles, or in special secretory cells and stored in cavities in the secreting glands (as in the leaves of mints, skin of oranges, etc.), or in special ducts (as in pines, milkweeds, etc.) or on the epidermis (as the "bloom" of plums, cabbages, etc., the resinous coating of many leaves, etc.). As a general rule, the glucosides, pigments, and enzymes are the products of unspecialized cells and have some definite connection with the metabolic processes of the plant; while the volatile oils and the alkaloids are usually secreted by special cells and have no known rôle in metabolism.[Pg 21]
CHAPTER III
PHOTOSYNTHESIS
Photosynthesis is the process whereby chlorophyll-containing plants, in the presence of sunlight, synthetize organic compounds from water and carbon dioxide. The end-product of photosynthesis is always a carbohydrate. Chemical compounds belonging to other groups, mentioned in the preceding chapter, are synthetized by plants from the carbohydrates and simple raw materials; but in such cases the energy used is not solar energy and the process is not photosynthesis.
Under the ordinary conditions of temperature, moisture supply, etc., necessary to plant growth, photosynthesis will take place if the three essential factors, chlorophyll, light, and carbon dioxide are available.
PHYSIOLOGICAL STEPS IN PHOTOSYNTHESIS
There are five successive and mutually dependent steps in the process of photosynthesis, as follows:
(1) There must be a gas exchange between the plant tissue and the surrounding air, by means of which the carbon dioxide of the air may reach the protoplasm of the chlorophyll-containing cells.
(2) Radiant energy must be absorbed, normally that of sunlight, although photosynthesis can be brought about by the energy from certain forms of artificial light.
(3) Carbon dioxide and water must be decomposed by the energy thus absorbed, and the nascent gases thus produced combined into some synthetic organic compound, with a resultant storage of potential energy.
(4) This first organic synthate must be condensed into some carbohydrate suitable for translocation and storage as reserve food.
(5) The oxygen, which is a by-product from the decomposition[Pg 22] of the water and carbon dioxide and the resultant synthetic process, must be returned to the air by a gas exchange.
Of the five steps in this process, the first two and the last are essentially purely physical phenomena, the chemical changes involved being those of the third and fourth steps. Hence, it is only these two parts of the process which need be taken into account in a consideration of the chemistry of photosynthesis.
FORMALDEHYDE, THE SIMPLEST CARBOHYDRATE STRUCTURE
The simplest carbohydrates known to occur commonly in plant tissues are the hexoses (see Chapter IV) having the formula C6H12O6, which is just six times that of formaldehyde, CH2O. Also, it is known that formaldehyde easily, and even spontaneously, polymerizes into more complex forms having the general formula (CH2O)n; trioxymethylene, C3H6O3, being a well-known example. Further, both trioxymethylene and formaldehyde itself can easily be condensed into hexoses, by simple treatment with lime water as a catalytic agent. Hence, it is commonly believed that formaldehyde is the first synthetic product resulting from photosynthesis, that this is immediately condensed into hexose sugars, and that these in turn are united into the more complex carbohydrate groups which are commonly found in plants (see Chapter IV).
There is considerable experimental confirmation of the soundness of this view. The whole photosynthetic process takes place in chlorophyll-containing plant tissues with astonishing rapidity, sugars, and even starch, appearing in the tissues almost immediately after their exposure to light in the presence of carbon dioxide. Hence, any intermediate product, such as formaldehyde, is present in the cell for only very brief periods and in very small amounts. But small amounts of formaldehyde can often be detected in fresh green plant tissues and, as will be pointed out below, the whole process of photosynthesis, proceeding through formaldehyde as an intermediate product, can be successfully duplicated in vitro in the laboratory.
Assuming, then, that formaldehyde is the first photosynthetic product in the process of the production of carbohydrates from water and carbon dioxide, the simple empirical equation for this transformation would be
H2O + CO2 = CH2O + O2. [Pg 23]
It is apparent, however, that the process is not so simple as this hypothetical reaction would indicate, as water and carbon dioxide can hardly be conceived to react together in any such simple way as this. Various theories as to the exact nature of the steps through which the chemical combinations proceed have been advanced. A discussion of the experimental evidence upon which these are based and of the conclusions which seem to be justified from these experimental studies is presented below. The only value which may be attached to the empirical equation just presented is that it does accurately represent the facts that a volume of oxygen, equal to that of the carbon dioxide consumed in the process, is liberated and that formaldehyde is the synthetical product of the reactions involved.
It should be noted, in this connection, that formaldehyde is a powerful plant poison and that few, if any, plant tissues can withstand the toxic effect of this substance when it is present in any considerable concentration. Hence, it is necessary to this whole conception of the relation of formaldehyde to the photosynthetic process, to assume that, however rapidly the formaldehyde may be produced in the cell, it is immediately converted into harmless carbohydrate forms.
THE CONDENSATION OF FORMALDEHYDE INTO SUGARS
As has been mentioned, it is easily possible to cause either formaldehyde, or trioxymethylene, to condense into C6H12O6, using milk of lime as a catalyst. Of course, no such condition as this prevails in the plant cell, and the mechanics of the protoplasmic process may be altogether different from those of the artificial syntheses. Furthermore, the hexose produced by the artificial condensation of these simpler compounds is, in every case, a non-optically active compound, while all natural sugars are optically active (see Chapter IV). Emil Fischer has succeeded, however, by a long and round-about process which need not be discussed in detail here, in converting the artificial hexose into glucose and fructose, the optically-active sugars which occur naturally in plant tissues. The condensation of formaldehyde directly into glucose and fructose in the plant cell is brought about by some process the nature of which is not yet understood. Probably synthetic enzymes (see Chapter XIV), whose nature[Pg 24] and action have not yet been discovered, come into play. It is a noteworthy fact, however, that the mechanics of this apparently simple chemical change, upon which the whole nutrition of the plant depends, and which furnishes the whole animal kingdom, including the human race, with so large a proportion of its food supplies, is as yet wholly unknown.
It is the common practice to represent the whole results of the photosynthetic action by the empirical equation
6H2O + 6CO2 = C6H12O6 + 6O2;
but here again the only value to be attached to such an algebraic expression is that it accurately represents the gaseous exchange of carbon dioxide and oxygen involved in the process. Certainly, it throws no light upon the nature of the process itself.
THEORIES CONCERNING PHOTOSYNTHESIS
The many theories which have been advanced concerning the nature of the chemical changes which are involved in photosynthesis have served as the basis for much experimental study of the problem. The following brief summary will serve to point out the general trend of these investigations and the present state of knowledge concerning the chemistry of photosynthesis.
Von Baeyer, in 1870, advanced the hypothesis that the first step in the process is the breaking down of carbon dioxide into carbon monoxide and oxygen and of water into hydrogen and oxygen; that the carbon monoxide and hydrogen then unite to produce formaldehyde, which is immediately polymerized to form a hexose. These theoretical changes may be represented by the following equations:
| 1. | CO2 = CO + O H2O = H2 + O |
| 2. | H2 + CO = CH2O |
| 3. | 6(CH2O) = C6H12O6 |
In the investigations and discussions of this hypothesis, it has been ascertained: first, that carbon monoxide has never been found in the free form in plant tissues; second, that when Tropaeolum plants were surrounded with an atmosphere in which there[Pg 25] was no carbon dioxide, but which contained sufficient carbon monoxide to give a concentration of this gas in the cell-sap equivalent to that in which CO2 is normally present, the plants grew normally and apparently elaborated starch; third, other and more extensive experiments indicated, however, that green plants in general cannot make use of carbon monoxide gas for photosynthesis, although this does not prove that von Baeyer's idea that CO is a step in the process is necessarily erroneous; and finally it was shown that carbon monoxide, in sufficient concentration to produce the results with Tropaeolum mentioned above, usually acts as a powerful anæsthetic towards most other plants. While these considerations do not positively prove that von Baeyer's hypothesis is incorrect, they render it so improbable that it has generally been abandoned in favor of others which are described below.
Erlenmeyer, even before the experimental work mentioned in the preceding paragraph had been reported, suggested that instead of assuming a separate breaking down of the carbon dioxide and water, it is easier to conceive that they are united in the cell-sap into carbonic acid and that this is reduced by the chlorophyll-containing protoplasm into formic acid and then to formaldehyde, as indicated by the following equations:
| 1. | H2CO3 = H2CO2 + O |
| 2. | H2CO2 = CH2O + O |
Like von Baeyer's hypothesis, this assumes that formaldehyde and oxygen are the first products of photosynthesis.
Proceeding upon this assumption, many investigators have studied the question as to whether formaldehyde actually is present in green leaves. Several workers have reported successful identification of formaldehyde in the distillate from green leaves; while others have criticized these results and have maintained that formaldehyde can likewise be obtained by distilling decoctions of dry hay, etc., in which the photosynthetic process could not possibly be conceived to be at work. Other investigators, notably Bach and Palacci, reported that they had succeeded in artificially producing formaldehyde from water and carbon dioxide, in the presence of a suitable catalyzer or sensitizer. Euler,[Pg 26] however, later showed conclusively that under the conditions described by these investigators, formaldehyde can be obtained even if no carbon dioxide is present, being apparently produced by the action of water upon the organic sensitizer which was used.
These conflicting reports led Usher and Priestley, in a series of studies reported between 1906 and 1911, to submit the whole matter to a critical review. Briefly, these investigators showed that the photolysis of carbon dioxide and water results in the formation of formaldehyde and hydrogen peroxide, as represented by the equation
CO2 + 3H2O = CH2O + 2H2O2.
The formaldehyde is then condensed by the protoplasm into sugars, while the hydrogen peroxide is decomposed, by an enzyme in the plant cell, into water and oxygen. If the formaldehyde is not used up rapidly enough by the protoplasm, it kills the enzyme and the undecomposed hydrogen peroxide destroys the chlorophyll, which stops the whole photosynthetic process. Usher and Priestley were able to cause the photolysis of carbon dioxide and water into formaldehyde outside of a green plant, in the presence of a suitable catalyzing agent which continually destroys the hydrogen peroxide as fast as it is formed; to show the actual bleaching effect of an excess of hydrogen peroxide in plant tissues which had been treated in such a way as to prevent the enzyme from decomposing it; and, finally, to demonstrate the condensation of formaldehyde into starch by the action of protoplasm which contained no chlorophyll.
In the meantime, Fenton, in 1907, found that in the presence of magnesium as a catalyst (it will be shown in Chapter VIII that magnesium is a constituent of the chlorophyll molecule) formaldehyde may be obtained from a solution of carbon dioxide in water, especially if weak bases are present.
Further, Usher and Priestley's later results showed that radium emanations, acting upon a solution of carbon dioxide in water, produce hydrogen peroxide and formaldehyde, and the latter polymerizes but not up to the point represented by the hexose sugars; also, that the ultra-violet rays from a mercury vapor lamp are very effective in bringing about the production of hydrogen peroxide and formaldehyde from a saturated aqueous[Pg 27] solution of carbon dioxide, the reaction taking place even in the absence of any "sensitizer," but much more readily if some "optical" or "chemical" sensitizer is present. Finally, these investigators were able to duplicate all their results, using green plant tissues, and to show that the temperature changes which take place in a film of chlorophyll when it is exposed to an atmosphere of moist carbon dioxide in the sunlight are such as would be required by the formation of formaldehyde and hydrogen peroxide from carbonic acid.
More recently, Ewart has showed that formaldehyde can combine chemically with chlorophyll; from which fact, Schryver deduces the theory that if for any reason the condensation of formaldehyde into carbohydrates by the cell protoplasm does not proceed as rapidly as the formaldehyde is produced by photosynthesis, the excess of the latter enters into combination with the chlorophyll, and that if condensation into sugar uses up all the free formaldehyde which is present in the active protoplasm, the compound of formaldehyde with chlorophyll is broken down setting free an additional supply for further sugar manufacture. According to this conception there are, in the chlorophyll-bearing protoplasm, not only the agencies for the production of formaldehyde from carbon dioxide and water and for the condensation of this into carbohydrates, but also a chemical mechanism by means of which the amount of free formaldehyde in the reacting mass may be regulated so that at no time will it reach the concentration which would be injurious to the cell protoplasm or fall below the proper proportions for sugar-formation. This explanation affords a satisfactory solution of the difficulty which formerly confronted the students of photosynthesis, namely, the fact that free formaldehyde is powerfully toxic to cell protoplasm. Without some such conception, it was difficult to imagine how the presence of formaldehyde in the cell contents, even as a transitory intermediate product, could be otherwise than injurious.
As a result of these studies, the nature of the chemical changes which result in the production of formaldehyde as the first product of photosynthesis, with the liberation of a volume of oxygen equal to that of the carbon dioxide consumed, seems to be fairly well established.[Pg 28]
THE PRODUCTION OF SUGARS AND STARCHES
The next step in the process, the conversion of formaldehyde into sugars and starches, is not necessarily a photosynthetic one, as it can be brought about by protoplasm which contains no chlorophyll or other energy-absorbing pigment. It is, however, a characteristic synthetic activity of living protoplasm. There is little definite knowledge as to how the cell protoplasm accomplishes this important task. As has been pointed out, the polymerization of formaldehyde into a sugar-like hexose, known as "acrose," can be easily accomplished by ordinary laboratory reactions, and acrose can be converted into glucose or fructose by a long and difficult series of transformations. But such processes as are employed in the laboratory to accomplish these artificial synthesis of optically-active sugars from formaldehyde can have no relation whatever to the methods of condensation which are used by cell protoplasm in its easy, almost instantaneous, and nearly continuous accomplishment of this transformation. Furthermore, these simple hexoses are by no means the final products of cell synthesis, even of carbohydrates alone. In many plants, starch appears as the final, if not the first, product of formaldehyde condensation. At least, the transformation of the simple sugars, which may be supposed to be the first products, into starch is effected so nearly instantaneously that it is impossible to detect measurable quantities of these sugars in the photosynthetically active cells of such plants. Other species of plants always show considerable quantities of simple sugars in the vegetative tissues, and some even store up their reserve carbohydrate food material in the form of glucose or sucrose. Attempts have been made to associate the type of carbohydrate formed in cell synthesis with the botanical families to which the plants belong, but with no very great success. For each individual species, however, the form of carbohydrate produced is always the same, at least under normal conditions of growth. For example, the sugar beet always stores up sucrose in its roots, although under abnormal conditions considerable quantities of raffinose are developed. Similarly, potatoes always store up starch, but with abnormally low temperatures considerable quantities of this may be converted into sugar, which becomes starch again with the return to normal conditions.[Pg 29]
While it is impossible, with our present knowledge, to even guess at the mechanism by which protoplasm condenses formaldehyde into sugars and these, in turn, into more complex carbohydrates, the structure and relationships to each other of the final products of photosynthesis are well known, and are discussed at length in the following chapter.
References
Barnes, C. R.—"Physiology" (Part II of Coulter, Barnes and Cowles' "Textbook of Botany"), 187 pages, 18 figs., Chicago, 1910.
Ganong, W. F.—"Plant Physiology," 265 pages, 65 figs., New York, 1908 (2d ed.).
Jost, L., trans. by Gibson, R. J. H.—"Plant Physiology," 564 pages, 172 figs., Oxford, 1907.
Marchlewski, L.—"Die Chemie des Chlorophylls," 187 pages, 5 figs., 7 plates, Berlin, 1909.
Parkin, John.—"The Carbohydrates of the Foliage Leaf of the Snowdrop (Galanthus nivalis L.) and their Bearing on the First Sugar of Photosynthesis," in Biochemical Journal, Vol. 6, pages 1 to 47, 1912.
Pfeffer, W., trans. by Ewart, A. J.—"Physiology of Plants." Vol. I, 632 pages, 70 figs., Oxford, 1900.
CHAPTER IV
CARBOHYDRATES
These substances comprise an exceedingly important group of compounds, the members of which constitute the major proportion of the dry matter of plants. The name "carbohydrate" indicates the fact that these compounds contain only carbon, hydrogen, and oxygen, the last two elements usually being present in the same proportions as in water. As a rule, natural carbohydrates contain six, or some multiple of six, carbon atoms and the same number of oxygen atoms less one for each additional group of six carbons above the first one; e.g., C6H12O6, C12H22O11, C18H32O16, etc.
Carbohydrates are classed as open-chain compounds, that is, they may be regarded as derivatives of the aliphatic hydrocarbons. From the standpoint of the characteristic groups which they contain, they are aldehyde-alcohols. In common with many other polyatomic open-chain alcohols, they generally possess a characteristic sweet, or mildly sweetish, taste. In the case of the more complex and less soluble forms, this sweetish taste is scarcely noticeable and these compounds are commonly called the "starches," as contrasted with the more soluble and sweeter forms, known as "sugars."
The characteristic ending ose is added to the names of the members of this group. As systematic names, the Latin numeral indicating the number of carbon atoms in the molecule is combined with this ending; e.g., C5H10O5, pentose, C6H12O6, hexose, etc.
In recent years, as a matter of scientific interest, many sugarlike substances which contain from two to nine carbon atoms combined with the proper number of hydrogen and oxygen atoms to be equivalent to the same number of molecules of water in each case, have been artificially prepared in the laboratory and designated as dioses, trioses, tetroses, pentoses, hexoses, heptoses,[Pg 31] octoses, and nonoses, respectively. Substances corresponding in composition and properties with the artificial tetroses and one or two derivatives of heptoses are occasionally found in plant tissues, and a considerable number of pentoses and their condensation products are common constituents of plant gums, etc.; but the great majority of the natural carbohydrates are hexoses and their derivatives.
GROUPS OF CARBOHYDRATES
Since the simpler carbohydrates are sugars, i.e., they possess the characteristic sweet taste, the name "saccharide" is used as a basis for the classification of the entire group. The simplest natural sugars, the hexoses, C6H12O6, are known as mono-saccharides. The group of next greater complexity, those which have the formula C12H22O11 and may be regarded as derived from the combination of two molecules of a hexose with the dropping out of one molecule of water at the point of union, are known as di-saccharides. Compounds having the formula C18H32O16 (i.e., three molecules of C6H12O6 minus two molecules of H2O) are tri-saccharides; and the still more complex groups, having the general formula (C6H10O5)n, are called the poly-saccharides. The mono-, di-, and tri-saccharides are generally easily soluble in water, have a more or less pronouncedly sweet taste, and are known as the sugars; while the polysaccharides are generally insoluble in water and of a neutral taste, and are called starches. As will be seen later, there are many natural plant carbohydrates belonging to each of these groups.
In addition to these saccharide groups, there are other types, or groups, of compounds which resemble the true carbohydrates in their chemical composition and properties and are often considered as a part of this general group. These are the pentoses, C5H10O5, and their condensation products, the pentosans (C5H8O4)n, and their methyl derivatives, C6H12O5; certain polyhydric alcohols having the formula C6H8(OH)6; pectose and its derivatives, pectin and pectic acid; and lignose substances of complex composition. It is doubtful whether these compounds are actual products of photosynthesis in plants, or have the same physiological uses as the carbohydrates and it has seemed wise to consider them in a separate and later chapter.[Pg 32]
ISOMERIC FORMS OF MONOSACCHARIDES
Four sugars having the formula C6H12O6, namely, glucose, fructose, mannose, and galactose, occur very commonly and widely distributed in plants. In addition to these, thirteen others having the same percentage composition have been artificially prepared, while seven additional forms are theoretically possible. In other words, twenty-four different compounds, all having the same empirical formula and similar sugar-like properties are theoretically possible. In order to arrive at a conception of this multiplicity of isomeric forms, it is necessary to understand the two types of isomerism which are involved. One of these is structural isomerism, and the other is space- or stereo-isomerism.
Structural Isomerism.—This refers to an actual difference in the characteristic groups which are present in the molecule. As has been said, all carbohydrates, from the standpoint of the characteristic groups which they contain, are aldehyde-alcohols. The hexoses all contain five alcoholic groups and one primary aldehyde, or one secondary aldehyde (ketone), group. If the aldehyde oxygen is attached to the carbon atom which is at the end of the six-membered chain, the structural arrangement is that of an aldehyde, 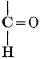 and the sugar is of the type known as "aldoses"; whereas, if the oxygen is attached to any other carbon in the chain, the ketone arrangement,
and the sugar is of the type known as "aldoses"; whereas, if the oxygen is attached to any other carbon in the chain, the ketone arrangement, 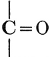 results and the sugar is a "ketose." This difference is illustrated in the Fischer open-chain formulas for glucose (an aldose) and fructose (a ketose) as follows:
results and the sugar is a "ketose." This difference is illustrated in the Fischer open-chain formulas for glucose (an aldose) and fructose (a ketose) as follows:
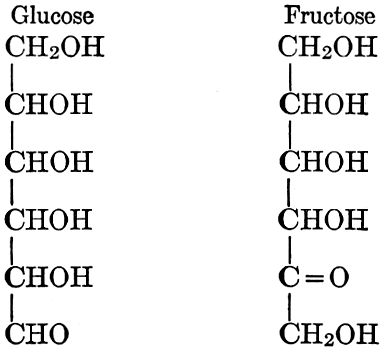
Stereo-isomerism, or space isomerism, as its name indicates, depends upon the different arrangement of the atoms or groups in the molecule in space, and not upon any difference in the character of the constituent groups. This possibility depends upon the existence in the molecule of the substance in question of one or more asymmetric carbon atoms and manifests itself in differences in the optical activity of the compound.[1] Thus, in the formula for glucose shown above there appear four asymmetric carbon atoms, namely, those of the four secondary alcohol groups (in the terminal, or primary alcohol, group, carbon is united to hydrogen by two bonds, and in the aldehyde group it is united to oxygen by two bonds). Similarly, fructose contains three asymmetric carbon atoms.
As an example of how the presence of these asymmetric carbon atoms results in the possibility of many different space relationships, the following graphic illustrations of the supposed differences between dextro-glucose and levo-glucose, and between dextro- and levo-galactose, may be cited.[2]
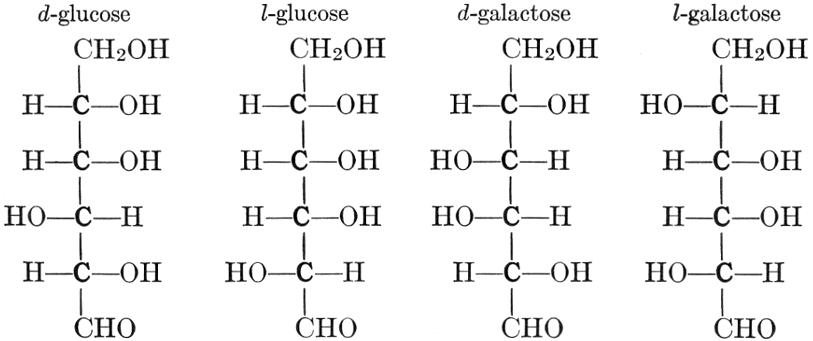
[Pg 34]Comparisons of the above formulas will show that the difference between the formulas for d- and l-glucose lies in the arrangement of the H atoms and the OH groups around the two asymmetric carbon atoms next the aldehyde end of the chain; while the d- and l-galactoses differ in that this arrangement is in the reverse order around all four of the asymmetric carbons. By similar variations in the grouping around the four asymmetric atoms, it is possible to produce the sixteen different space arrangements shown on page 37 for the groups of an aldohexose. Sugars corresponding to fourteen of these different forms have been discovered, three of which are of common occurrence in plants, either as single mono-saccharides or as constituent groups in the more complex carbohydrates; the remaining two forms have only theoretical interest.[Pg 35]
Similarly, for a ketohexose, which contains three asymmetric carbon atoms, there are eight possible arrangements. Three sugars of this type are known, only one (fructose) being common in plants; the others are of only theoretical interest.
FOOTNOTES:
[1] It is assumed that the reader, or student, is familiar with the theoretical and experimental evidence in support of the existence of the so-called "asymmetric" carbon atom and its relation to the effect of the compound which contains it, when in solution, in rotating the plane of polarized light. For purposes of review, or of study of this most interesting and important phenomenon, the reader is referred to any standard text-book on Organic Chemistry.
[2] Attention should be called, at this point, to the fact that such formulas as these cannot possibly accurately represent the actual arrangement of the constituent groups of a carbohydrate molecule around an asymmetric carbon atom. The limitations of a plane-surface formula prevent any illustration of the three-dimension relationships in space. Furthermore, there are certain facts in connection with the birotation phenomenon and the relation of the molecular configuration to biochemical properties (which see) that cannot be explained on the basis of the open-chain arrangement represented by the Fischer formulas used here. A closed-ring arrangement, showing the aldehyde oxygen as linked by its two bonds to the first and the fourth carbon atoms of the chain, thus forming a closed-ring of four carbon and one oxygen atoms, instead of being attached by both bonds to a single carbon atom, as in the above formulas, is undoubtedly a more nearly accurate representation of the actual linkage in the molecule than are the open-chain formulas used above.
The differences in conception embodied by these two types of formulas may be shown by the following formulas for glucose:
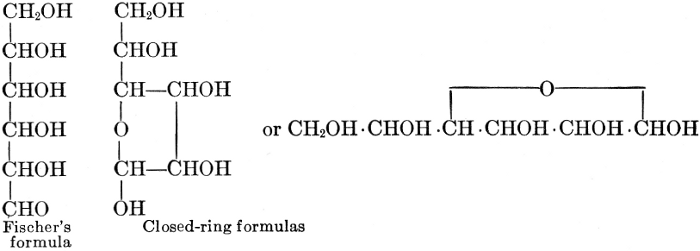
It will be observed that in the closed-ring formula there are five asymmetric carbon atoms, and the asymmetry of the terminal one forms the basis for the explanation of the existence of the so-called α and β modification of d-glucose (see page 46). However, the ordinary aldehyde reactions of the sugars are more clearly indicated by the open-chain formula. Some investigators are inclined to be that sugars actually exist in the open-chain arrangement when in aqueous solution, and in the closed-ring arrangement when in alcoholic solution. The closed-ring formulas will be used in this text in the discussions of the birotation phenomena and of biochemical properties, but for the explanations of the stereo-isomeric forms and similar phenomena, the open-chain formulas are just as useful in conveying an idea of the possibilities of different space relationships, and are so much simpler in appearance and in mechanical preparation, that it seems desirable to use these rather than the more accurate closed-ring formulas.
CHEMICAL CONSTITUTION OF MONOSACCHARIDES
The term "monosaccharides," as commonly used, refers to hexoses. It applies equally well, however, to any other sugar-like substance which either occurs naturally or results from the decomposition of more complex carbohydrates, and which cannot be further broken down without destroying its characteristic aldehyde-alcohol groups and sugar-like properties.
All such monosaccharides, being alcohol-aldehydes, can easily be reduced to the corresponding polyatomic alcohols, containing the same number of carbon atoms as the original monosaccharides, each with one OH group attached to it. All aldose monosaccharides are converted, by gentle oxidation, into the corresponding monobasic acid, having a COOH group in the place of the original CHO group. Further oxidation either changes the alcoholic groups into COOH groups, producing polybasic acids, or breaks up the chain. When ketose monosaccharides are submitted to similar oxidation processes, they are broken down into shorter chain compounds.
The various monosaccharides which have thus far been found as constituents of plant tissues, or as parts of other more complex compounds which occur in plants, are shown in the following table:[Pg 36]
| Trioses (C3H6O3) | Tetroses (C4H8O4) | |||
| Aldose— | Glyceric aldehyde, or glycerose | Aldoses— | d- and l-Erythrose, | |
| l-Threose | ||||
| Ketose— | Dioxyacetone | |||
| Pentoses (C5H10O5) | Methyl Pentoses (C6H12O5) | |||
| Aldoses— | d- and l-Arabinose | Aldoses— | Rhamnose | |
| d- and l-Xylose | Fucose | |||
| l-Ribose | Rhodeose | |||
| l-Lyxose | Chinovose | |||
| Hexoses (C6H12O6) | ||||
| Mannitol series | Dulcitol series | |||
| Aldoses— | d- and l-Glucose | d- and l-Galactose | ||
| d- and l-Mannose | d- and l-Talose | |||
| d- and l-Gulose | ||||
| d- and l-Idose | ||||
| d-Altrose | ||||
| d-Allose | ||||
| Ketoses— | d-Fructose | d-Tagatose | ||
| d-Sorbose | ||||
| Heptoses (C7H14O7) | Octoses (C8H16O8) | Nonoses (C9H18O9) |
| Glucoheptose | Gluco-octose | Glucononose |
| Mannoheptose | Manno-octose | Mannononose |
| Galactoheptose | Galacto-octose | |
| Persuelose | ||
| Sedoheptose |
The hexoses are by far the most important group of monosaccharides. They are undoubtedly the first products of photosynthesis, and all the other carbohydrates may be considered to be derived from them by condensation. Because of their biochemical significance and their immense importance as the fundamental substances for all plant and animal energy-producing materials, the following detailed studies of their chemical composition and molecular configuration are fully warranted.
That all the hexoses contain five alcoholic groups is proved by the experimental evidence that each one forms a penta-ester, by uniting with five acid radicals, when treated with mineral or organic acids under proper conditions. Thus, glucose penta-acetate, penta-nitrate, penta-benzoate, etc., have all been prepared. The presence of the aldehyde group is proved by the fact that all aldohexoses have been converted, by gentle oxidation, into pentaoxy-monobasic acids, and the ketohexoses broken down into shorter chain compounds by similar gentle oxidations; these reactions being characteristic of compounds containing an aldehyde and a ketone group respectively. This experimental evidence establishes the nature of the characteristic groups in the molecule, in each case.
The molecular configurations illustrated in the following table are those suggested by Emil Fischer, as a result of his exhaustive[Pg 37] studies of the chemical constitution of the various carbohydrates. There is, of course, no thought that the printed formulas here presented accurately represent the actual relationships in space of the different groups; but there is fairly conclusive evidence that the variations in special groupings in the different sugars are properly referable to the particular asymmetric carbon atoms as indicated in the several formulas as presented.
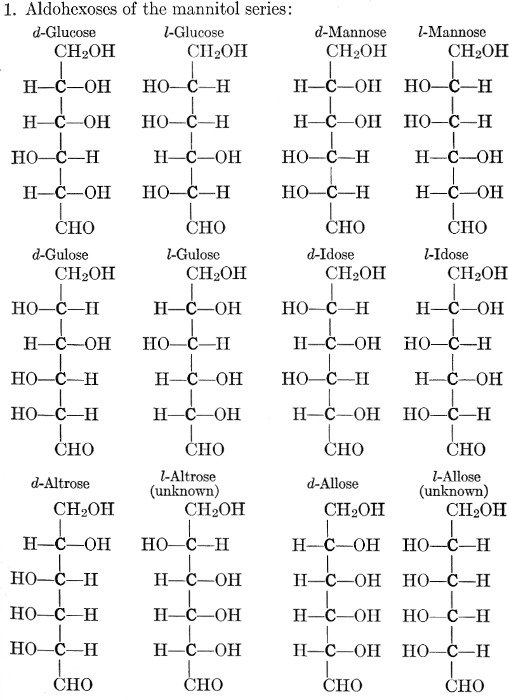
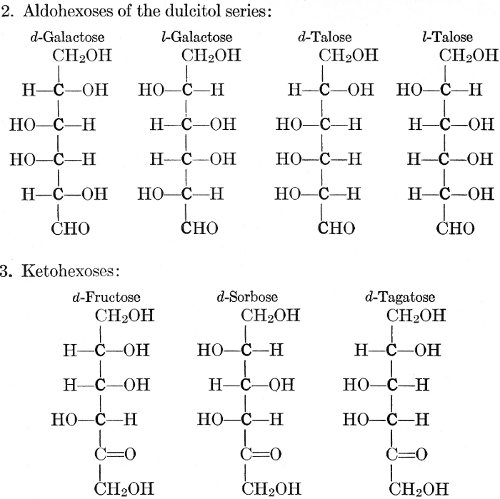
Reference will be made in subsequent paragraphs to the probable chemical constitution of the monosaccharides other than hexoses; but the above discussion of the structure of the hexoses will serve as a sufficient introduction to the study of the composition of the common carbohydrates.
CHARACTERISTIC REACTIONS OF HEXOSES
Specific Rotatory Power.—All soluble carbohydrates, since they contain asymmetric carbon atoms, with the consequent larger groups on one side of the molecule than the other, rotate the plane of polarized light when it passes through a solution of the carbohydrate in question. The amount of the rotation depends upon the nature of the carbohydrate, the concentration of the solution, and the length of the column of solution through which the ray of polarized light passes. But the same definite amount[Pg 39] of the same sugar, dissolved in the same volume of water, and placed in a tube of the same length, will always cause the same angular deviation, or rotation, of the plane in which the polarized light which passes through it is vibrated. In other words, the same number of molecules of the optically active substance in solution will always produce the same rotatory effect. This is called the specific rotatory power of the substance in question. It is expressed as the number of degrees of angular deviation of the plane of polarized light caused by a column of the solution exactly 200 mm. in length, the concentration of the solution being 100 grams of substance in 100 cc. at a temperature of 20° C. Actual determinations of specific rotatory power are usually made with solutions more dilute than this standard, and the observed deviation multiplied by the proper factor to determine the effect which would be produced by the solution of standard concentration. If the direction of the deviation is to the right (i.e., in the direction in which the hands of the clock move) it is spoken of as "dextro" rotation and is indicated by the sign +, or the letter d; while if in the opposite direction, it is called "levo" rotation and indicated by the sign -, or the letter l. For example, the specific rotation of ordinary glucose is +52.7°; of fructose, -92°; of sucrose, +66.5°.
Reducing Action.—All of the hexose sugars are active reducing agents. This is because of the aldehyde group which they contain. Many of the common heavy metals, when in alkaline solutions, are strongly reduced when boiled with solutions of the hexose sugars. Alkaline copper solutions yield a precipitate of red cuprous oxide; ammoniacal silver solutions give silver mirrors; alkaline solutions of mercury salts are reduced to metallic mercury, etc. Any sugar which contains a potentially active aldehyde group will exhibit this reducing effect and is known as a "reducing sugar." In some of the di- and tri-saccharides, the linkage of the hexose components together is through the aldehyde group, in such a way that it loses its reducing effect; such sugars are known as "non-reducing." Advantage is taken of this property for both the detection and quantitative determination of the "reducing sugars." A standard alkaline copper solution of definite strength, known as "Fehling's solution," is added to the solution of the sugar to be tested and the mixture boiled, when the characteristic brick-red precipitate appears. If certain standard[Pg 40] conditions of volume of solutions used, length of time of boiling, etc., are observed, the quantity of cuprous oxide precipitated bears a definite ratio to the amount of sugar which is present, so that if the precipitate be filtered off and weighed under proper conditions, the weight of sugar present in the original solution can be calculated. The proper conditions for carrying on such a determination and tables showing the amounts of the various "reducing sugars" which correspond to the weight of cuprous oxide found, are given in all standard text-books dealing with the analysis of organic compounds.
Fermentability.—The common hexoses are all easily fermented by yeast, forming alcohol and carbon dioxide, according to the equation
C6H12O6 = 2C2H5OH + 2CO2.
The importance and biochemical significance of this reaction will be considered in detail in connection with the discussions of the relation of molecular configuration to biochemical properties (see page 56) and the nature of enzyme action (see page 194).
Formation of Hydrazones and Osazones.—Another property of the hexoses which is due to the presence of an aldehyde group in the molecule, is that of forming addition products with phenyl hydrazine, known as "hydrazones" and "osazones." For example, glucose reacts with phenyl hydrazine in acetic acid solution, in two stages. The first, which takes place even in a cold solution may be represented by the equation
| C6H12O6 | + | C6H5·NH·NH2 | = | C6H12O5:N·NH·C6H5 | + | H2O. |
| Glucose | Phenyl-hydrazine | Glucose-hydrazone |
The structural relationships involved may be represented as follows:
The hydrazones of the common sugars, with the exception of the one from mannose, are colorless compounds, easily soluble in[Pg 41] water. Hence, they do not serve for the separation or identification of the individual sugars. But if the solution in which they are formed contains an excess of phenyl hydrazine and is heated to the temperature of boiling water for some time, the alcoholic group next to the aldehyde group (the terminal alcohol group in ketoses) is first oxidized to an aldehyde and then a second molecule of phenyl hydrazine is added on, as illustrated above, forming a di-addition-product, known as an "osazone." The osazones are generally more or less soluble in hot water, but on cooling they crystallize out in yellow crystalline masses each with definite melting point and crystalline form. All sugars which have active aldehyde groups in the molecule form osazones. These afford excellent means of identification of unknown sugars, or of distinguishing between sugars of different origin and type.
Glucose, mannose, and fructose all form identical osazones. This is because the structure of these three sugars is identical except for the arrangement within the two groups at the aldehyde end of the molecule (see formulas on page 44). Since it is to these two groups that the phenyl hydrazine residue attaches itself, it follows that the resulting osazones must be identical in structure and properties. All other reducing sugars yield osazones of different physical properties.
When an osazone is decomposed by boiling with strong acids, the phenyl hydrazine groups break off, leaving a compound containing both an aldehyde and a ketone group. Such compounds are known as "osones." The osones from glucose, mannose, and fructose are identical. By carefully controlled reduction, either one of the C=O groups of the osone may be changed to an alcoholic group, producing thereby one of the original sugars again. Hence, it is possible to start with one of these sugars, convert it into the osone and then reduce this to another sugar, thereby accomplishing the transformation of one sugar into another isomeric sugar.
Formation of Glucosides.—By treatment with a considerable variety of different types of compounds, under proper conditions, it is possible to replace one of the hydrogen atoms of the terminal alcoholic group of the hexose sugars with the characteristic group of the other substance, forming compounds known, respectively, as glucosides, fructosides, galactosides, etc. The structural relation of methyl glucoside to glucose, for example, may be illustrated as follows:[Pg 42]

A general formula for glucosides is R·(CHOH)5·CHO; and the R may represent a great variety of different organic radicals (see the chapters dealing with Glucosides and with Tannins). When the glucosides are hydrolyzed, they yield glucose and the hydroxyl compound of the radical with which it is united. All the statements which have been made with reference to glucosides, apply equally well with reference to fructosides, galactosides, mannosides, etc.
It is possible, by various laboratory processes, to replace additional hydrogen atoms in the glucose molecule with the same or other organic radicals, thus producing glucosides containing two or more R groups; but most of the natural glucosides contain only one other characteristic group.
Oxidations.—When the hexoses are oxidized they give rise to three different types of acids, depending upon the conditions of the oxidation and the kind of oxidizing agent used. With glucose, for example, the relationships involved may be illustrated as follows:
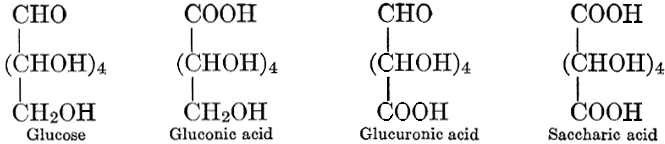
An important property of the acids of the gluconic type is that when heated with pyridine or quinoline to 130°-150° they undergo a molecular rearrangement whereby the acid corresponding to an isomeric sugar is produced. For example, gluconic acid, under these conditions, becomes mannonic acid, which can be reduced to mannose. The process is reversible; mannose can be converted to mannonic acid, thence to gluconic acid, thence to glucose. Similarly, galactonic acid can be converted into talonic acid, and this to talose, and this process is reversible. These facts afford another means of conversion of one sugar into another.[Pg 43]
From the standpoint of physiological processes, glucuronic acid is the most interesting and important oxidation product of glucose. It is often found in the urine of animals, as the result of the partial oxidation of glucose in the animal tissues. Normally, glucose is oxidized in the body to its final oxidation products, carbon dioxide and water. But when many difficultly oxidizable substances, such as chloral, camphor, turpentine oil, aniline, etc., are introduced into the body, the organism has the power of combining these with glucose to form glucosides. These so-called "paired" compounds are then oxidized to the corresponding glucuronic acid derivatives and eliminated from the body in the urine. No phenomenon similar to this occurs in plants, however, and glucuronic acid has never been found in plant tissues.
Synthesis and Degradation of Hexoses.—Monosaccharides of any desired number of carbon atoms can be produced from aldoses having one less carbon atoms, by way of the familiar "nitrile" reaction. Aldoses, like all other aldehydes, combine directly with hydrocyanic acid, forming compounds known as nitriles, which contain one more carbon atom than was present in the original aldehyde; the cyanogen group can easily be converted into a COOH group; and this, in turn, reduced to an aldehyde, thus producing an aldose with one more carbon atom than was present in the initial sugar. These changes may be illustrated by the following equations:
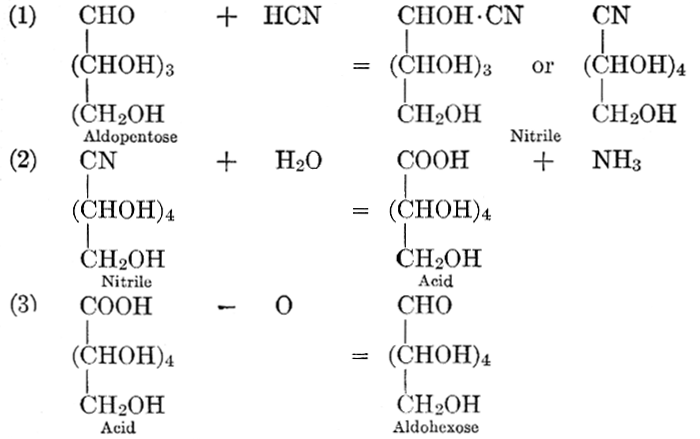
It is possible, by this process, to advance step by step from formaldehyde to higher sugars, Emil Fischer and his students[Pg 44] having carried the process as far as the production of glucodecose (C10H20O10). It usually happens, however, that two stereo-isomers result from the "step-up" by way of the nitrile reaction; thus, arabinose yields a mixture of glucose and mannose, glucose yields glucoheptose and mannoheptose, etc.
The reverse process, or the so-called "degradation" of a sugar into another containing fewer carbon atoms, may be readily accomplished in either one or two ways. In Wohl's process, the aldehyde group of the sugar is first converted into an oxime, by treatment with hydroxylamine; the oxime, on being heated with concentrated sodium hydroxide solution, splits off water and becomes the corresponding nitrile; this, on further heating, splits off HCN and yields an aldose having one less carbon atom than the original sugar. This process is the exact reverse of the nitrile synthesis, described above. The second method of degradation, suggested by Ruff, makes use of Fenton's method of oxidizing aldehyde sugars to the corresponding monobasic acid, using hydrogen peroxide and ferrous sulfate as the oxidizing mixture; the aldonic acid thus formed is then converted into its calcium salt, which, when further oxidized, splits off its carboxyl group and one of the hydrogens of the adjacent alcoholic group, leaving an aldose having one less carbon atom than the original aldose sugar.
Enolic Forms.—A final avenue for the interconversion of glucose, mannose, and fructose into one another, is through the spontaneous transformations which these undergo when dissolved in water containing sodium hydroxide or potassium hydroxide. This change is due to the conversion of the sugar, in the alkaline solution, into an enol, which is identical for all three sugars, and which may subsequently be reconverted into any one of the three isomeric hexoses. The relationships involved are illustrated in the following formulas:
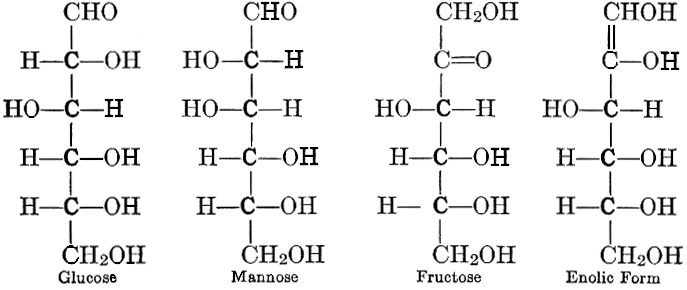
The preceding technical discussion of the chemical constitution and reactions of the hexoses has been presented, not because it has any direct connection with the occurrence or functions of these compounds in plant tissues, but for the purpose of giving to the student a graphic conception of the structure and properties of these simple carbohydrates, as a basis for the understanding of the nature, properties, possible chemical reactions, syntheses, etc., of the more complex types of carbohydrates, which, along with these simple monosaccharides, constitute the most important single group of organic components of plants.
THE OCCURRENCE AND PROPERTIES OF MONOSACCHARIDES
Only two monosaccharides occur as such in plants. These are glucose and fructose. All the other hexoses, whose structure is shown on pages 37 and 38, occur in plants only as constituents of the more complex saccharides, in glucoside-formations, or as the corresponding polyatomic alcohols.
The aldo-hexoses which occur most commonly in plants, either free or in combination, are d-glucose, d-mannose, and d-galactose; while d-fructose and d-sorbose are the common keto-hexoses.
Glucose (often called also dextrose, fruit sugar, or grape sugar) occurs widely distributed in plants, most commonly in the juices of ripening fruits, where it is usually associated with fructose and sucrose, the two hexoses being easily derived from sucrose by hydrolysis. Glucose is also produced by the hydrolysis of many of the more complex carbohydrates, by the action either of enzymes or of dilute acids; lactose, maltose, raffinose, starch, and cellulose, as well as many glucosides all yielding glucose as one of the products of their hydrolysis. In all such cases, it is d-glucose which is obtained.
Glucose is a crystalline solid (although it does not form such sharply defined crystals as does sucrose, or "granulated sugar"), which is easily soluble in water. It usually appears on the market in the form of thick syrups, which are produced commercially by the hydrolysis of starch with dilute sulfuric acid, removal of the acid after the hydrolysis is complete, and evaporation of the resulting solution to the desired syrupy consistency. (Since corn starch is commonly used as the raw material for this process, these syrups are often spoken of as "corn syrup.") The sweetness of glucose is about three-fifths that of ordinary cane sugar.[Pg 46]
Glucose exhibits all the properties of hexoses which have been described in general terms above. It is a reducing-sugar, and is easily fermented. The specific rotatory power of d-glucose is +52.7°. But when glucose is dissolved in water, it exhibits in a marked degree the phenomenon known as "mutarotation"; that is, freshly made solutions exhibit a certain definite rotatory power, but this changes rapidly until it finally reaches another definite specific rotation. In other words, glucose is "birotatory," or possesses two distinct specific rotatory powers, and the changing rotation effect in aqueous solutions is due to the change from one form to the other. When dissolved in alcohol, it does not exhibit this change in rotatory power. In order to explain this phenomenon, it is necessary to assume that there are two modifications of d-glucose, which have been designated respectively as the α and β forms. The possibility of the existence of these two forms is explained by the assumption of the closed-ring arrangement of the glucose molecule, as indicated in the following formulas which represent the two possible isomeric arrangements:
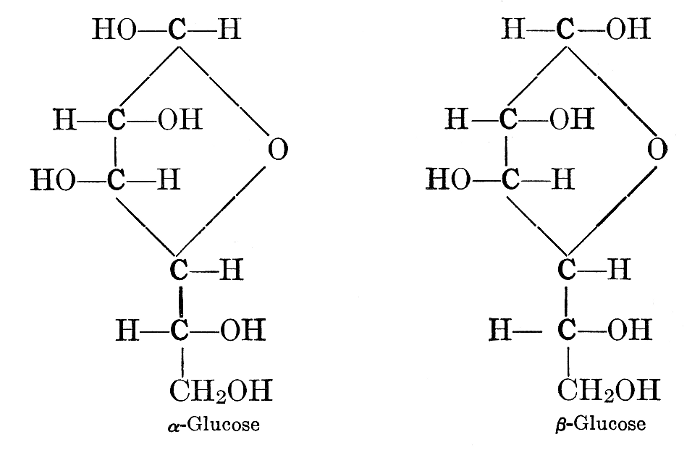
It is assumed that the α modification (with its specific rotatory power of +105°) is the normal form for crystalline glucose, but that when dissolved in water it is changed into an aldehydrol, i.e., a compound containing two additional OH groups, which later breaks down again, into the β modification (with its specific rotatory power of +22°). When dissolved in alcohol, this change does not take place because of the absence of the excess of water necessary to produce the intermediate aldehydrol form.[Pg 47]
There are other examples of the existence of the α and β modification of glucose. For example, α-methyl-glucoside and β-methyl-glucoside (specific rotatory powers, +157° and -33°, respectively) are both known, as well as several other similar glucoside arrangements.
Mannose.—This sugar does not occur as such in plants; but complex compounds which yield d-mannose when hydrolyzed, known as "mannosans," are found in a number of tropical plant forms. The mannose which is obtained from these by hydrolysis is very similar to glucose in its properties, forms the same osazones as do glucose and fructose, exhibits mutarotation, etc. Mannose may also be obtained by oxidizing mannitol, a hexatomic alcohol, known as "mannite," which occurs in many plants, especially in the manna-ash (Fraxinus ornus), the dried sap from which is known as "manna."
Galactose occurs in the animal kingdom as one of the constituents of lactose, or milk-sugar. It is also one of the constituents of raffinose, a trisaccharide sugar found in plants, and occurs as "galactans" in many gums and sea-weeds. The d-galactose, obtained by the hydrolysis of any of these compounds, is a faintly sweet substance which resembles glucose in many of its properties; having one characteristic difference, however, in that it forms mucic acid instead of saccharic acid when oxidized by concentrated nitric acid. These oxidation products are very different in their physical properties and this difference serves to distinguish between the two sugars from which they are derived.
Fructose (levulose, honey sugar, or "diabetic" sugar) occurs along with glucose in the juices of many fruits, etc. It is a constituent of sucrose, of raffinose, and of the polysaccharide inulin, from which it may be obtained by hydrolysis. It is a ketose sugar, reduces Fehling's solution, forms the same osazone as glucose, and is easily fermentable by yeast. Its sweetness is slightly greater than that of ordinary cane sugar. d-fructose (the ordinary form) is easily soluble in water, and is strongly levorotatory, its specific rotatory power at 20° C. being -92.5°; it is unique in the very large effect which is produced in its rotatory power by increasing the temperature of the solution; at 87° its specific rotatory power is reduced to -52.7°, exactly equal to but in the opposite direction of the effect of glucose; hence, invert sugar, which is a mixture of an equal number of molecules of glucose and fructose, and which[Pg 48] has a specific rotatory power of -19.4° at 20° C., becomes optically inactive at 82° C.
Sorbose is the only other ketohexose which has any importance in plant chemistry. It does not occur free in plants, but is the first oxidation product from the hexatomic alcohol, sorbitol, which is present in the juice of the berries of the mountain-ash. Sorbose is a crystalline solid, which is not fermentable by yeast, but which otherwise closely resembles fructose.
DISACCHARIDES
The disaccharides, having the formula C12H22O11, may be regarded as derived from the monosaccharides by the linking together of two hexose groups with the dropping out of a molecule of water, in the same way that many other organic compounds form such linkages. That this is a perfectly correct conception, is shown by the fact that, when hydrolyzed, the disaccharides break down into two hexose sugars, thus
C12H22O11 + H2O = C6H12O6 + C6H12O6.
With all known disaccharides, at least one of the hexoses obtained by hydrolysis is glucose; hence all disaccharides may be regarded as glucosides (C6H12O5·R) in which the R is another hexose group.
Since hexoses have both alcoholic and aldehyde groups, and since either of these types of groups may function in the linkage of the two hexoses to form a disaccharide, it is possible for two hexoses, both of which are reducing sugars to be linked together in three different ways: (1) through an alcoholic group of each hexose, (2) through an alcoholic group of one and the aldehyde group of the other, and (3) through the aldehyde group of each hexose. Disaccharides linked in either of the first two ways will be reducing sugars, since they still contain a potentially active aldehyde group; but those of the third type will not be reducing sugars, since the linkage through the aldehyde groups destroys their power of acting as reducing agents. Examples of each of these three types of linkage are found among the common disaccharides, as will be pointed out below.
The following table shows the general characteristics of the common disaccharides.[Pg 49]
| Type 1.—Aldehyde group potentially active, reducing sugars: | |
| Sugar | Components |
|---|---|
| Maltose | Glucose and glucose |
| Gentiobiose | Glucose and glucose |
| Lactose | Glucose and galactose |
| Melibiose | Glucose and galactose |
| Turanose | Glucose and fructose |
| Type 2.—Non-reducing sugars: | |
| Sucrose | Glucose and fructose |
| Trehalose | Glucose and glucose |
The disaccharides of Type 1 reduce Fehling's solution and form hydrazones and osazones, although somewhat less readily than do the hexoses. They all show mutarotation and exist in two modifications, indicating that the component groups have the closed-ring arrangement.
The disaccharides of Type 2, since they contain no potentially active aldehyde group, do not reduce Fehling's solution, nor form osazones; neither do they exhibit mutarotation. The only disaccharides which occur as such in plants are of this type. Disaccharides of Type 1 may be obtained by the hydrolysis of other, more complex, carbohydrates.
All disaccharides are easily hydrolyzed into mixtures of their component hexoses, by boiling with dilute mineral acids, or by treatment with certain specific enzymes which are adapted to the particular disaccharide in each case (see page 55, also Chapter XIV).
Sucrose (cane sugar, beet sugar, maple sugar) is the ordinary "granulated sugar" of commerce. It occurs widely distributed in plants, where it serves as reserve food material. It is found in largest proportions in the stalks of sugar cane, in the roots of certain varieties of beets, and in the spring sap of maple trees, all of which serve as industrial sources for the sugar. In the sugar cane, and beet-roots, it constitutes from 12 to 20 per cent of the green weight of the tissue and from 75 to 90 per cent of the soluble solids in the juice which can be expressed from it. Its universal use as a sweetening agent is due to the combined facts that it crystallizes readily out of concentrated solutions and, hence, can be easily manufactured in solid form, and that it is sweeter than any other of the common sugars except fructose.[Pg 50]
Sucrose is a non-reducing sugar, forms no osazone, and is not directly fermentable by yeast, although most species of yeasts contain an enzyme which will hydrolyze sucrose into its component hexoses, which then readily ferment.
When hydrolyzed by acids, or by the enzyme "invertase," it yields a mixture of equal quantities of glucose and fructose. Sucrose is dextrorotatory, but since fructose has a greater specific rotatory action to the left than glucose has to the right, the mixture resulting from the hydrolysis of sucrose is levorotatory. Since the hydrolysis of sucrose changes the rotatory effect of the solution from the right to the left, the process is usually called the "inversion" of sucrose, and the resultant mixture of equal parts of glucose and fructose is called "invert sugar." As has been pointed out, solutions of invert sugar become optically inactive when heated to 82 °C., because of the reduction in the rotatory power of fructose due to the higher temperature.
The probable linkage of the two hexoses to form sucrose, in such a way as to produce a non-reducing sugar, is illustrated in the following formula:
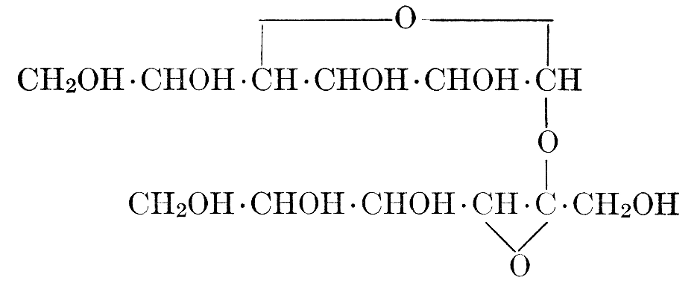
Trehalose seems to serve as the reserve food for fungi in much the same way that sucrose does for higher plants. It is composed of two molecules of glucose linked together through the aldehyde group of each, as trehalose is a non-reducing sugar. This linkage is illustrated in the following formula:
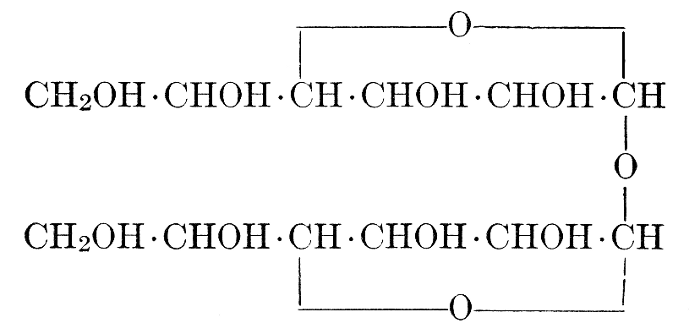
Trehalose may be hydrolyzed into glucose by dilute acids and by the enzyme "trehalase," which is contained in many yeasts and in several species of fungi. It is strongly dextrorotatory (specific rotatory power, +199°). It is not fermentable by yeast.
Trehalose appears to replace sucrose in those plants which contain no chlorophyll and do not elaborate starch. The quantity of trehalose in such plants reaches a maximum just before spore formation begins. Since it is manufactured in the absence of chlorophyll, its formation must be accomplished by some other means than photosynthesis, yet it is composed wholly of glucose—a natural photosynthetic product.
Maltose rarely occurs as such in plants, although its presence in the cell-sap of leaves has sometimes been reported. It is produced in large quantities by the hydrolysis of starch during the germination of barley and other grains. This hydrolysis is brought about by the enzyme "diastase," which is present in the sprouting grain.
Maltose is easily soluble in water, and crystallizes in masses of slender needles. It is a reducing sugar; readily forms a characteristic osazone; is strongly dextrorotatory (specific rotatory power +137°); and is readily fermented by ordinary brewer's yeast, which contains both "maltase" (the enzyme which hydrolyzes maltose to glucose) and "zymase" (the alcohol-producing enzyme). When hydrolyzed, either by dilute acids or by maltase, one molecule of maltose yields two molecules of glucose. Its component hexoses are, therefore, the same as those of trehalose, a non-reducing sugar, this difference in properties being due to the difference in the point of linkage between the two glucose molecules, that for maltose being such as to leave one of the aldehyde groups potentially active, as shown in the following formula,
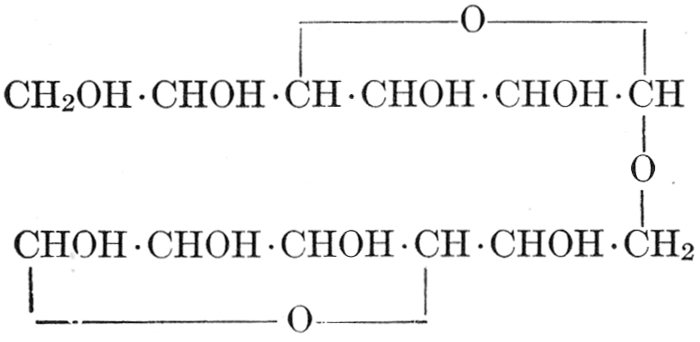
Isomaltose is a synthetic sugar, obtained by Fischer, by condensing two molecules of glucose. Its properties are quite similar to those of maltose, but it yields a slightly different osazone and is not fermentable by yeast. These differences are explained by the assumption that this sugar is a glucose-β-glucoside, while normal maltose is a glucose-α-glucoside.
Gentiobiose is a disaccharide which results from the partial hydrolysis of the trisaccharide gentianose (see page 53). It is very similar in its general properties to isomaltose. Cellobiose is a disaccharide which results from the hydrolysis of cellulose. It is a reducing sugar, forms an osazone, and resembles maltose.
Maltose, isomaltose, gentiobiose, and cellobiose, are all glucose-glucosides, the difference between them being undoubtedly due to linkage being between different alcoholic groups in the glucose molecules.
The disaccharide lactose is a glucose-galactoside. It is the sugar which is present in the milk of all mammals. It has never been found in plants. Melibiose, which is the corresponding vegetable glucose-galactoside, may be obtained by the partial hydrolysis of the trisaccharide raffinose (see below). It is a reducing sugar; forms a characteristic osazone; and exhibits mutarotation. It is not fermented by ordinary top-yeasts, but is first hydrolyzed and then fermented by the enzymes present in bottom-yeasts.
TRISACCHARIDES
Trisaccharides, as the name indicates, consist of three hexoses (or monosaccharides) linked together by the dropping out of two molecules of water. Their formula is C18H32O16. When completely hydrolyzed, they yield three molecules of monosaccharides; when partially hydrolyzed, one each of a disaccharide and a monosaccharide.
One trisaccharide of the reducing sugar type, namely rhamnose, exists in plants as a constituent of the glucoside xanthorhamnin. It is composed of one molecule of glucose united to two molecules of rhamnose (methyl pentose, C6H12O5). It is of interest only in connection with the properties of the glucoside in which it is present (see page 84).
Three trisaccharides which are non-reducing sugars are found in plants; namely, raffinose, gentianose, and melizitose.[Pg 53]
Raffinose occurs normally in cotton seeds, in barley grains, and in manna; also, in small quantities in the beet root, associated with sucrose. It is more soluble in water than is sucrose and hence remains in solution in the molasses from beet-sugar manufacture, which constitutes the commercial source for this sugar. Raffinose crystallizes out of concentrated solutions, with five molecules of water of crystallization, in clusters of glistening prisms. It is strongly dextrorotatory, the anhydrous sugar having a specific rotatory power of +185°, and the crystalline form, C18H32O16, showing a specific rotation of +104.5°. It does not reduce Fehling's solution, nor form an osazone, and in its other properties it closely resembles sucrose.
The hydrolysis of raffinose presents several interesting possibilities. If its structure is represented as follows:
C6H11O5——C6H10O4——C6H11O5
Fructose Glucose Galactose
\_____ _____/ \_____ _____/
\/ \/
Sucrose Melibiose
it is apparent that it may break down by hydrolysis in three different ways: (1) into sucrose and galactose, (2) into fructose and melibiose, and (3) into fructose, glucose, and galactose. As a matter of fact, it does actually break down in these three different ways, under the influence of different catalysts; invertase or dilute acids break it down into fructose and melibiose, emulsin hydrolyzes it to sucrose and galactose, while strong acids or the enzymes of bottom-yeasts break it down into the three hexoses.
Gentianose, a trisaccharide found in the roots of yellow gentian (Gentiana lutea), is a non-reducing sugar, which when hydrolyzed yields either fructose and gentiobiose, or fructose and two molecules of glucose.
Melizitose, a trisaccharide which, in crystallized form, has the formula, C18H32O16·2H2O, occurs in the sap of Larix europea and in Persian manna, and has recently been found in considerable quantities in the manna which collects on the twigs of Douglas fir and other conifers. When hydrolyzed, it yields one molecule of fructose and one of turanose, a disaccharide containing fructose and glucose linked together in a slightly different way than they are in sucrose. Turanose itself is a reducing sugar, but when linked with fructose to form melizitose its reducing properties are destroyed. Melizitose is a very sweet sugar.[Pg 54]
TETRASACCHARIDES
A complex saccharide, known as stachyose, which is found in the tubers of Stachys tuberifera, is said by some investigators to be a tetrasaccharide and by others to have the formula C36H62O31·7H2O (i.e., a hexasaccharide). It is a crystalline solid, with a faintly sweetish taste, and a specific rotatory power of +148°. When hydrolyzed it yields glucose, fructose, and two (or more) molecules of galactose.
THE RELATION OF THE MOLECULAR CONFIGURATION OF SUGARS TO THEIR BIOCHEMICAL PROPERTIES
As will be pointed out later (see Chapter XIV), all chemical reactions which are involved in vital phenomena, including those of plant growth and metabolism, are controlled by enzymes. The biochemical reactions which the soluble carbohydrates undergo afford such excellent illustrations of the relation of the molecular configuration of an organic compound to the possibility of the action of an enzyme upon it, that it seems desirable to discuss this relationship at this point, rather than to postpone it until after the nature of enzyme action has been considered. Undoubtedly, the student, after he has studied the nature of enzymes and their mode of action, as presented in Chapter XIV, will find it profitable to return to this section and review the facts here presented, as illustrating the principles and mechanism of enzyme action. But a consideration, at this time, of the relation of the molecular configuration of the sugars to their biochemical reactions cannot fail to add interest to the study of these matters from the chemical and biological standpoints.
It has been known for a long time that the dextro- and levo-isomers of a compound which contains one or more asymmetric carbon atoms are affected differently by biological agents, such as yeasts, moulds, bacteria, etc. Pasteur, as early as 1850, showed that the green mould, Penicillium glaucum, when growing in solutions of racemic acid (a mixture of equal molecules of d- and l-tartaric acids) uses up only the d-acid, leaving the l-form absolutely untouched. Later, it was found that the same green mould attacks l-mandelic acid in preference to the d- form; whereas the[Pg 55] yeast, Saccharomyces ellipsoideus, exhibits the opposite preference for these acids.
These observations upon some of the earlier known forms of optically active organic acids led the way to a general study of this phenomenon as exhibited by the optically active soluble carbohydrates. The results of these studies may be considered in connection with the several different types of reactions which these sugars undergo, as follows:
Glucoside Hydrolysis.—As was pointed out in connection with the discussion of the mutarotation of glucose, this sugar may exist in either the α or the β modification. Glucosides of both α and β glucose are of common occurrence. The difference in molecular configuration, in such cases, may be represented by the following formulas:
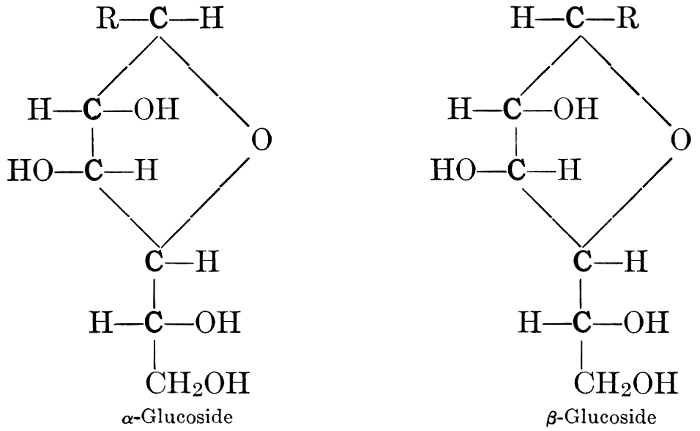
The radical represented by the R may be either a common alkyl radical (as CH3, C2H5, etc.), another saccharide group (as in the case of the disaccharides, trisaccharides, etc.), or some other complex organic group (as in the case of the natural glucosides described in Chapter VI). But, in every case, the glucoside is easily hydrolyzed by the enzyme maltase (or α-glucase) if the molecular arrangement is that represented by the α-attachment, or by the enzyme emulsin (or β-glucase) if the glucoside is of the β type; but emulsin is absolutely without effect upon α-glucosides, and maltase does not produce the slightest change in β-glucosides. These statements hold true regardless of the nature of the group which is represented by the R in the formulas above. Hence, the[Pg 56] biochemical properties of the glucosides, so far as their hydrolysis by the enzymes which are present in many biological agents is concerned, depends wholly upon the molecular configuration of the glucose itself. Furthermore, neither the mannosides, which differ from glucosides only in the arrangement of the H and OH groups attached to one of the asymmetric carbon atoms in the hexose, nor galactosides in which two such arrangements are different (see configuration formulas on page 57), are attacked by either maltase or emulsin. But other enzymes specifically attack other disacharides, or polysaccharides, or glucoside-like complexes. For example, lactase acts energetically upon ordinary lactose and all other β-galactosides; but not upon any glucoside, mannoside, etc.
Again, neither α- nor β-xylosides, which correspond with the above-described glucosides in every particular except that the HCOH group next the terminal CH2OH group is missing, are hydrolyzed by either emulsin or maltase.
These instances, selected from among many similar observations, clearly prove that not only the number and kind of groups in the molecule, but also the arrangement of the constituent groups in space, must be identical in order that the compound may be acted upon by any given enzyme acting as a biological hydrolytic agent.
Fermentability.—The enzyme zymase, present in all yeasts, promotes the fermentation of the natural d- forms of the three hexoses, glucose, mannose, and fructose, but is without effect upon the artificial l- forms of the same sugars. The uniform action of zymase upon these hexoses is easily explained upon the basis of the same assumption which was used to account for the formation of identical osazones from these sugars and their easy transformation into each other; namely, their easy transformation into an enolic form which is identical for all three.
Further, galactose is fermented by some yeasts (although not by all), but much less readily than are the other sugars, and the temperature reaction is quite different with galactose than with the others. Talose and tagatose are entirely unfermentable. A study of the configuration formulas for these several sugars shows the explanation for these observed facts. These formulas are as follows:[Pg 57]
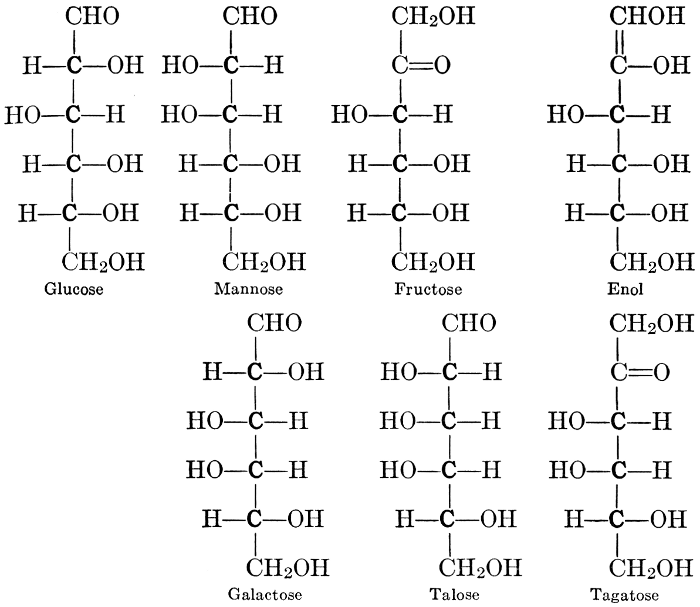
It will be noted that in the case of glucose, mannose, and fructose, the configuration is identical at every point except at the aldehyde end of the chain, and that here the two groups readily arrange themselves into the same enolic form for the three sugars. Galactose differs from these three sugars only in the arrangement of the H and OH groups attached to one of the other carbon atoms (the third from the alcoholic end); the difficulty of its fermentation indicates that some molecular rearrangement to bring this group into its proper configuration must precede the fermentation process. The fact that it is the third HCOH group which thus undergoes rearrangement is significant because of the participation of these parts of molecules in groups of threes in many biological processes, as will be mentioned elsewhere. Talose is unfermentable, even though the arrangement of its upper three groups is the same as in the galactose and the lower three the same as in mannose.
If further proof that fermentability depends upon molecular configuration were needed, it is furnished by the fact that no pentose is fermentible, even though the stereo-arrangement of[Pg 58] each of the four alcoholic groups in the molecule is identical with the corresponding groups in a fermentible hexose.
Oxidation by Bacteria.—The bacillus Bacterium xylinum contains an enzyme, or enzymes, which promote the oxidation of the aldehyde group of an aldose sugar to COOH, or of one alcoholic CHOH group next the terminal CH2OH group of a hexatomic alcohol to C=O. But these oxidizing enzymes affect only those compounds in which the OH groups are on the same side of the two asymmetric carbon atoms next the end of the molecule where the oxidation takes place, as indicated in the following groupings.
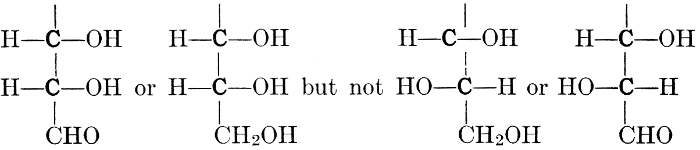
The configuration of the remainder of the molecule is immaterial to action by these oxidizing bacteria; hence, the enzymes in this case are apparently concerned only with the configuration arrangement of a portion of the molecule, instead of with the whole hexose grouping, as in the cases of the other reactions which have been thus far considered.
It is apparent from these illustrations, and from many more which might be cited, that there is a very definite relation between the molecular configuration of a carbohydrate and its biochemical properties, as represented by the possibilities of the action of enzymes upon it. The probable nature of this relationship will be better understood after the general questions involved in the mode of enzyme action have been considered (see Chapter XIV). But for the present, it will be sufficient to note that it seems to be necessary that the enzyme shall actually fit the molecular arrangement of the compound at all points, in the same way that a key fits its appropriate lock; or a still better illustration is that of the fitting of a glove to the hand. On the basis of the latter illustration, it is just as impossible for a dextro-enzyme to affect a levo-sugar, or for α-glucase to affect a β-glucoside, as it is to fit a right-hand glove upon a left hand. Further attention will be given to these matters in later chapters.[Pg 59]
POLYSACCHARIDES
The polysaccharides which, like the simpler saccharides, or sugars, which have thus far been studied, undoubtedly serve as reserve food for plants, are known under the general name of "starches." They are substances of high molecular weight, whose constitution is represented by the general formula (C6H10O5)n. It should be noted that an exactly accurate formula should be (C6)n(H12O6)n-1; but since the value of n is very high, the simpler formula is approximately correct. The value of n has not been accurately determined for any of the individual members of the group, but is probably never less than 30 and may often be 200 or more. The fact that these compounds are insoluble in most of the solvents which can be used for molecular weight determinations makes it difficult to determine their actual molecular constitution.
When completely hydrolyzed, the polysaccharides yield only hexoses. They are, therefore, technically known as "hexosans." Each individual polysaccharide which has been studied thus far yields only a single hexose, although the particular hexose obtained varies in different cases. In fact, the polysaccharides are often classified according to the hexoses which they yield on hydrolysis, into the following groups: the dextrosans, which yield glucose, and include starch, dextrin, glycogen, lichenin, etc.; the levulosans, which yield fructose, and include inulin, graminin, triticin, etc.; the mannans; and the galactans. The more common representatives of each of these groups are discussed below.
(A) The Dextrosans
These are by far the most common type of polysaccharides to be found in plants.
Starch.—It is probable that no other single organic compound is so widely distributed in plants as is ordinary starch. It is produced in large quantities in green leaves as the temporary storage form of photosynthetic products. As a permanent reserve food material, it occurs in seeds, in fruits, in tubers, in the pith, medullary rays and cortex of the stems of perennials, etc. It constitutes from 50 to 65 per cent of the dry weight of seeds of cereals, and as high as 80 per cent of the dry matter of potato tubers.[Pg 60]
Starch occurs in plant tissues in the form of microscopic granules, composed of concentric layers, there being apparently alternate layers of two types of carbohydrate material, which have been distinguished from each other by several different pairs of names used by different authors: thus, Nägeli uses the terms "granulose" and "amylocellulose"; Meyer, "α and β amylose"; Wolff, "amylo-cellulose" and "amylo-pectin"; while Kramer asserts that the layers are alternate lamella of crystalline and colloidal starch. Many theories as to the nature of these concentric layers and their mode of deposition have been advanced, but it would not be profitable to discuss them in detail here.
For purposes of study, starch may be prepared from the ground meal of cereals, potatoes, etc., by kneading the meal in a bag or sieve of fine-meshed muslin or silk, under a slow stream of water. The starch granules, being microscopic in size, readily pass through the cloth with the water, and may be caught in any suitable container. The starch is then allowed to settle to the bottom, the water poured off and the starch collected and dried.
Starch is insoluble in water; but if boiled in water, the granules burst and a slimy opalescent mass, known as "starch paste," is obtained. This is undoubtedly a colloidal suspension of the starch in water. By various processes, such as boiling with very dilute acids, treatment with acetone, etc., starch is converted into "soluble starch" which dissolves in water to a clear solution. Soluble starch is precipitated out of solution by alcohol, or by lead subacetate solution.
Air-dried starch contains from 15 to 20 per cent of water; but this can be completely removed, without altering the starch in any way, by heating for some time at 100° C.
The starch granules from different sources vary considerably in size and shape, and can generally be identified by observation under the microscope.
The most characteristic reaction of starch is the blue color which it gives with iodine. The reaction is most marked with starch paste or soluble starch, but even dry starch granules are colored blue when moistened with a solution of iodine in water containing potassium iodide, or with tincture of iodine.
When hydrolyzed, either by boiling with dilute acids or under the influence of enzymes, starch undergoes a series of decompositions, yielding first dextrins, then maltose, and finally glucose.[Pg 61] These transformations can be traced by the iodine color reaction, as starch will show its characteristic blue, dextrins purple or rose-red, and maltose and glucose no color with iodine.
Dextrins may occur in plants as transition products in the transformation of starch into sugars, or vice versa. Most commonly, however, they are artificial products resulting from the partial hydrolysis of starch in the laboratory or factory. They are amorphous substances, which are readily soluble in water, forming sticky solutions which are often used as adhesives ("library paste" is a common example of a very concentrated preparation of this kind). They are precipitated from solution by alcohol, but not by lead subacetate (distinction from starch). They are strongly dextrorotatory (specific rotatory power +192° to +196°); are not fermented by yeast alone, but readily undergo hydrolysis to glucose which does ferment. There are several different modifications, or forms, of dextrins, depending upon the extent to which the simplification of the starch molecule by hydrolysis is carried. Three fairly definite forms are generally recognized, as follows: amylo-dextrin, or soluble starch, slightly soluble in cold water, readily so in hot water, giving a blue color with iodine; erythro-dextrin easily soluble in water, neutral taste, red color with iodine; and achroo-dextrin, easily soluble in water, sweetish taste, no color with iodine.
Commercial dextrin, which is much used in the preparation of mucilages and adhesive pastes, is prepared by heating dry starch to about 250° C. It is composed chiefly of achroo-dextrin, mixed with varying quantities of erythro-dextrin and glucose.
Glycogen, or "animal starch," is one of the most widely distributed reserve foods of the animal body; in fact, it is the only known form of carbohydrate-reserve in animal tissues. But it is present only rarely in plants. It occurs in certain fungi, particularly in yeasts. In the animal body, glycogen is found in all growing cells; also in the muscles and blood; but most largely in the liver, where it is stored in large quantities. The glycogen found in yeasts is identical with that found in animal tissues. The quantity of glycogen in a yeast cell increases rapidly as the yeast grows during the fermentation process.
Glycogen is a white, amorphous compound, readily soluble in hot water, forming an opalescent solution similar in appearance to the solutions of soluble starch. It is strongly dextrorotatory[Pg 62] (specific rotatory power +190°), is colored brown by iodine, and is hydrolyzed to dextrin and maltose, and finally to glucose.
Lichenin, para dextran, and para isodextran are dextrosans which have been isolated from various lower plants. They all yield glucose when completely hydrolyzed. They resemble starch in chemical properties, but differ from it in physical form, etc.
(B) Levulosans
Inulin replaces starch as the reserve food carbohydrate in a considerable number of natural orders of plants, particularly in the Compositae. It is the carbohydrate of the tubers of the dahlia and artichoke and of the fleshy roots of chicory. It is often found associated with starch in monocotyledonous plants, such as many species of Iris, Hyacinthus, and Muscari. Among the monocotyledons, starch seems to be the characteristic carbohydrate reserve of aquatic, or moisture-loving, species, while inulin is more common among those which prefer dry situations.
Inulin may be prepared from the tubers of dahlias or artichokes, by boiling the crushed tubers with water containing a little chalk (to precipitate mineral salts, albumins, etc.) filtering and cooling the filtrate practically to the freezing point, which precipitates the inulin.
Inulin is a white, tasteless, semi-crystalline powder, which is soluble in hot water, from which it may be precipitated by alcohol or by freezing. It forms no paste like that of starch or dextrin, and gives no color with iodine. It is levorotatory, and when hydrolyzed by acids or by the enzyme inulinase yields fructose; in fact, inulin bears the same relation to fructose that starch does to glucose.
Graminin, irisin, phlein, sinistrin, and triticin are all inulin-like polysaccharides, which have been found in the plants after which they are named. Their solutions are, as a rule, sticky or gummy in consistency, which suggests that these compounds bear the same relation to inulin that dextrins do to starch.
(C) Mannosans, or Mannans
Mannan bears the same relation to mannose that starch does to glucose and inulin to fructose. It occurs as a reserve food sub[Pg 63]stance in many plants. It has been reported as present in moulds, and in ergot; in the roots of asparagus, chicory, etc.; in the leaves and wood of many trees, such as the chestnut, apple, mulberry, and many conifers; also as a part of the so-called "hemi-celluloses" which are present in the seeds of many plants, notably the palms, the elders, cedar, larch, etc.
It is a white, amorphous powder, which is difficultly soluble in water, is strongly dextrorotatory (specific rotatory power +285°), and when hydrolyzed yields mannose.
Secalin (or carubin) is a substance which is found in the seeds of barley, rye, etc., which is similar to mannan, but is optically inactive.
(D) Galactans
These bear the same relation to galactose that the preceding dextrosans do to their constituent hexoses. Four different galactans have been isolated from plant tissues; they are all white, amorphous solids which dissolve with difficulty in water, forming gummy solutions.
Both galactans and mannans commonly occur associated with cellulose and hemi-celluloses in the seeds or other storage organs of plants. They are practically indigestible by animals, as the proper enzymes to hydrolyze them are not present in the digestive tract; hence, they are commonly classed with the indigestible cellulose as the "crude fiber" of plants which are to be used as food by animals.
PHYSIOLOGICAL USE AND BIOLOGICAL SIGNIFICANCE OF CARBOHYDRATES
If the organic compounds produced by plants be classified with reference to their uses in metabolism into the three groups known, respectively, as temporary foods, storage products, and permanent structures, it is clear that the carbohydrates which have been discussed in this chapter may fall into either one of the first two of these classes. There can be no doubt that the first products of photosynthesis, whichever ones they may be in different plants, may be directly used as temporary foods, to furnish the energy and material for the building up of permanent structures. Also, there can be no doubt that these same carbohydrates are trans[Pg 64]located to the storage organs and accumulated for later use by the same plant (as, for example, in the case of the perennials), or by the next generation of the plant (when the storage is in the endosperm adjoining the embryo of the seed).
There is no known explanation as to why different species of plants make use of different carbohydrates for these purposes; or why certain species elaborate starch out of the same raw materials from which other species produce sugars, inulin, or glycogen, etc.
In general, starch is the final product of photosynthesis in most green plants; but there are many exceptions to this. The polysaccharides, which are generally insoluble, must be broken down into the simpler soluble sugars before they can be translocated to other organs of the plant for immediate, or future, use. When they reach the storage organs, they may be recondensed into insoluble polysaccharides, or stored as soluble sugars. Examples of the latter type of storage are, sucrose in beet roots, glucose in onion bulbs, etc. Sometimes, this habit of storage seems to be a species characteristic; as potatoes store starch, while beets, growing in the same soil and under exactly the same environment, store sugar. But in other cases, the nature of the carbohydrate stored undoubtedly is correlated with the external temperatures at the time of storage. It has been shown that cold, which tends to physiological dryness, very frequently favors the storage of sugars instead of starches. Thus, in temperate zones, among aquatic, or moisture-loving plants, those species which hibernate during the winter at the bottom of lakes or ponds and are killed by temperatures below freezing, store starch and no sugar; while in the same ponds, the species whose storage organs pass the winter above the level of the water and can withstand temperatures as low as -7° C. contain sugar during the winter months, even if they contain starch during warmer periods. Similarly, sugars often appear in the leaves and stems of conifers during the winter months, only to disappear, or be replaced by starch, when spring approaches. This same phenomenon is noticeable in arctic plants, which generally contain but small proportions of starch and relatively large amounts of sugars.
Similarly, the phenomenon of the turning sweet of potatoes when exposed to low temperatures has often been noted. The change of the starch in potato tubers to sugar is most rapid at the tempera[Pg 65]ture of 0° C., and ceases at 7°, or above. Also, if potatoes in which the maximum amount of sugar is present (not over one-sixth of the total starch can be converted into sugar) are exposed to a higher temperature the sugar soon disappears.
In general, however, it may be said that each particular species of plant has its own particular preference for a specific carbohydrate as its reserve food material, and elaborates the proper enzymes to make it possible to utilize this particular carbohydrate for its metabolic needs.
Again, the question as to whether the storage of energy-producing materials for the use of the next generation shall be in the form of carbohydrates or of fats seems to be definitely connected with the size of the seed, and the consequent available storage space (see page 138). Animals habitually use the space-conserving form of fats for their energy-storage, while plants more commonly use carbohydrates for this purpose, except in the case of those small seeds in which sufficient energy cannot be stored in carbohydrate form to develop the young seedling to the point where it can manufacture its own food. As a general rule, nuts, which contain the embryo of slow-growing seedlings, and need large proportions of energy reserve, are characteristically oily instead of starchy in type.
But, aside from temperature reactions and space requirements, there is no law which has yet been discovered which determines the character of the energy-storage compound which any given species of plant will elaborate. The process of photosynthesis would seem to be identical in all cases, at least up to the point of the production of the first hexose sugar; but the transformation of glucose into other monosaccharides, disaccharides, and polysaccharides seems to be a matter which obeys no rule or law.
Finally, there remains to be considered the occurrence and uses of sugars in the fleshy tissues of fruits. These tissues have, of course, no direct function in the life history of the plant. They surround the seed, but they must decay or be destroyed before the seed can come into the proper environment for germination and growth. In most fruits, starch is the form in which the carbohydrate material is first deposited in the green tissue, but as the fruit ripens the starch rapidly changes into sugars, with the result that the fruit takes on a flavor which makes it much more attractive as a food for men and animals. This purely biological significance[Pg 66] of the presence of sugars (and of the other substances which give desirable flavors to fruits, vegetables, etc.), can have no possible relation to the physiological needs of the individual plant, however.
It is apparent that the production of these immense stores of reserve food by plants makes them useful as food for animals, and it is, of course, the storage parts of the plants which are most useful for this purpose. This biological relationship needs no further emphasis.
REFERENCES
Abderhalden, E.—"Biochemisches Handlexikon, Band 2 ... Die Einfachen Zuckerarten, Inuline, Cellulosen, ...," 729 pages, Berlin, 1911, and "Band 8—1 Ergänzungsband (same title as Band 2)—" 507 pages; Berlin, 1914.
Armstrong, E. F.—"The Simple Carbohydrates and Glucosides," 233 pages. Monographs on Biochemistry, London, 1919 (3d ed.).
Fischer, E.—"Untersuchung ueber Kohlenhydrate und Fermente, 1884-1908," 912 pages, Berlin, 1909.
Mackensie, J. E.—"The Sugars and their Simple Derivatives," 242 pages, 17 figs., London, 1913.
Tollens, B.—"Kurzes Handbuch der Kohlenhydrate", 816 pages, 29 figs., Leipzig, 1914 (3d ed.).
CHAPTER V
GUMS, PECTINS, AND CELLULOSES
These substances constitute a group of compounds which are very similar to the polysaccharide carbohydrates in composition and constitution, but which serve entirely different purposes in the plant. As a class, they are condensation products of pentoses, known as pentosans and having the formula (C5H8O4)n, or hexosans having the formula (C6H10O5)n, or combined pentosan-hexosans.
In general, these compounds make up the skeleton, or structural framework material, of the plant, in contrast with the protoplasmic materials or food substances for which most of the other types of organic compounds (discussed in other chapters of this book) serve. They are the principal constituents of "woody fiber," of cell-walls, and of the "middle lamella" which fills up the spaces between the plant cells. They are, therefore, found in largest proportions in the stems of woody plants; but they are also present in every other organ of plants, as the cell-wall or other structural material.
For purposes of study, these compounds may conveniently be divided into three groups; namely, the natural gums and pentosans, the pectins and mucilages, and the celluloses. The segregation into these three groups is not sharply defined. The distinction between the groups is based upon the solubility of the compounds in water. The gums and pentosans readily dissolve in water; the pectins form colloidal solutions which are easily converted into "jellies"; the mucilages do not dissolve but form slimy masses; while the celluloses are insoluble in and unaltered by water. Some authors add a fourth group, known as "humins"; but as these are the products of decay (usually in the soil) of these structural compounds, rather than of growth and development, they need not be taken into consideration in a study of the chemistry of plant growth.[Pg 68]
THE NATURAL GUMS AND PENTOSANS
The natural gums, when hydrolyzed, yield large proportions of sugars, but most of them also contain a complex organic acid nucleus, by means of which they form salts with calcium, magnesium, etc. Some of them, such as cherry gum and those which are found in the woody stems of plants (wood gum, and those found in corn stalks, the straw of cereals, etc.) yield practically pure pentoses. These are known as pentosans. They bear the same relation to the pentose sugars as do the dextrosans to glucose, etc. The wound gums, for example, yield arabinose, and the wood gums yield xylose. But most of the natural gums yield a mixture of galactose, some pentose, and some complex organic acid.
The gums are translucent, amorphous substances, whose solutions in water are levorotatory. They are precipitated out of solution by alcohol and by lead subacetate solution.
Gums are extremely difficult to hydrolyze, the laboratory process of hydrolysis usually requiring from eighteen to twenty-four hours of continuous boiling with acids for its completion. Because of this difficulty of hydrolysis, gums are practically indigestible by animals and of little use as food.
The following common examples will serve to illustrate the general nature of these compounds.
Gum arabic, found in the exudate from the stems of various species of Acacia, is a mixture of the calcium, magnesium, and potassium salts of a diaraban-tetragalactan-arabic acid. Arabic acid has the formula C23H38O22, and one molecule of this acid serves as the nucleus for the union of eight galactose and four arabinose groups, linked together in some unknown way. The formula for the compound, exclusive of the metallic elements with which it is loosely united is C91H150O78. This gives some idea of its complexity.
When boiled with nitric acid, it is oxidized to mucic, saccharic, and oxalic acids. It gives characteristic reactions with alum, basic lead acetate, and other common reagents.
Gum arabic comes on the market as a brittle, glassy mass, which is used in the preparation of mucilages, and as a carrier for essential oils, etc., in certain toilet preparations.
Recent investigations have shown that the so-called "meta-pectic acid," which is often found in sugar beets and interferes[Pg 69] with the process of sugar manufacture, is identical with gum arabic in composition and properties.
Gum tragacanth is the soluble portion of the natural gum which is found in several species of Astragalus. It constitutes only 8 to 10 per cent of the total gum-like material which is present, the remainder being composed of insoluble gummy substances of unknown composition. The soluble gum consists of calcium, potassium, and magnesium salts of an acid which, when hydrolyzed, yields several molecules of arabinose, six of galactose, and one of geddic acid (an isomer of arabic acid). It is said to be produced by the metamorphosis of the medullary rays under unfavorable conditions of growth. It comes on the market in globular masses of amorphous material, and is used in the manufacture of cosmetics, etc.
Wound gum is frequently found in the tracheæ of plants, and near surface wounds, which it stanches. It is secreted by the cells surrounding the injured part. It responds to the reactions of other gums and to some of those of woody fiber. Its exact composition is not known, but probably lies between that of the true gums and that of cellulose.
These gums are generally considered to be decomposition products of celluloses, resulting from the action of some hydrolytic ferment, usually stimulated by some unfavorable condition of growth, some injury, or some morbid condition.
The pentosans, araban and xylan, occur normally in the stems and outer seed coats of many common plants. They constitute a considerable proportion of these tissues, as indicated by the following results of typical analyses: Wheat bran, 22 to 25 per cent; clover hay, 8 to 10 per cent; oat straw, 16 to 20 per cent; wheat straw, 26 to 27 per cent; corn bran, 38 to 43 per cent; jute fiber, 13 to 15 per cent; various wood gums, 60 to 92 per cent.
They are white, fluffy solids, which are difficultly soluble in cold water, more readily in hot water. They are very difficult to hydrolyze, and indigestible by animals. When finally hydrolyzed, they yield arabinose and xylose, respectively. The pith of dry corn stalks is a good illustration of their general character.[Pg 70]
MUCILAGES
These are characterized by forming slimy masses when moistened with water. They are secreted by hairs on the skin of many plants, so that the external walls of the leaves, fruit, and seeds are often mucilaginous when damp. This is particularly true of aquatic plants. The chemical composition of the mucilages is unknown. When hydrolyzed, they yield arabinose and a hexose; the latter is sometimes galactose and sometimes mannose.
When present on the surface of plant tissues, the mucilages probably serve to prevent the too rapid diffusion of materials through the skin, in the case of the aquatic plants, and too rapid transpiration, in the case of young vegetative tissues or in other plants when growing under extremely dry conditions. When found in tubers, or other storage organs, it has been supposed that they may serve as reserve food materials, but it seems that such difficultly hydrolyzable compounds as these can hardly function as normal reserve foods.
PECTINS
Many fruits, such as currants, gooseberries, apples, pears, etc., and many fleshy roots of vegetables, such as carrots, parsnips, etc., contain substances known as pectins. These are readily soluble in water, and when dissolved in concentrated solutions in hot water, they set into "jellies" when the solution is cooled. These jellies carry with them the soluble sugars and flavors which are present in the fruits, and constitute a familiar article of diet.
There are undoubtedly several different modifications of the pectins, to which the names "meta-pectin," "para-pectin," "pectic acid," "meta-pectic acid," and "para-pectic acid," have been applied. These all seem to be products of hydrolysis of a mother substance known as "pectose," which constitutes the middle lamella of unripe fruit, etc. As the fruit ripens, the pectose is hydrolyzed into the various semi-acid, or acid, bodies mentioned above. The intermediate products of the hydrolysis are the pectins, which swell up in water and readily form jellies; while the final meta-pectic acid is easily soluble in water and resembles the true gums in its properties. When the middle lamella reaches the pectic acid stage, the fruit becomes soft and "mushy" in texture.[Pg 71]
The pectins more nearly approach to the composition, properties, and functions of the celluloses than do any of the other groups of organic compounds. They have been extensively studied in connection with the parasitism of certain fungous diseases which cause the soft rots of fruits and vegetables. These parasites usually penetrate the tissues of the host plant by dissolving out the middle lamella material, which may sometimes serve as food material for the fungus; but more often the parasite secures its food supply from the protoplasm of the cell contents. In such cases, the parasite secretes both a pectose-dissolving enzyme, known as "pectase" and a "cellulase" which attacks the cell-wall material in order to provide for the entry of the fungus into the cells. Other enzymes, known as "pectinases," which coagulate the soluble pectins or pectic acids into insoluble jellies in the tissues of the plants seem to aid the plant in resisting the penetration by the parasite.
CELLULOSES
Used in its general sense, this term includes all those substances which are elaborated by protoplasm to constitute the cell-wall material. Cellulose proper is a definite chemical compound, whose properties are well established. In plants, however, this true cellulose is nearly always contaminated by various encrusting materials; and in the process of wood-formation, the cell-wall material continually thickens by the conversion of the cellulose into ligno-cellulose and the protoplasm of the cell as continuously diminishes in volume. Thus the protoplasm of the cell produces a number of different kinds of material which are deposited in the walls of the cell. All of these, taken together, constitute the general group known as the celluloses.
These may be divided into three classes: namely, (1) the hemi-celluloses, (2) the normal celluloses, and (3) the compound celluloses.
The hemi-celluloses (pseudo-, or reserve celluloses) include a series of complex polysaccharides which occur in the cell-walls of the seeds of various plants. They are found in the shells of nuts, rinds of cocoanuts, shells of stony fruits, etc., and in the seedcoats of beans, peas and other legumes. They are much more easily hydrolyzed than the other members of this group, and when[Pg 72] hydrolyzed yield various sugars, chiefly galactose, mannose, and the pentoses. They bear the same relation to these sugars that starch does to glucose, and are generally supposed to serve as reserve food material, although it is difficult to conceive how the shells, etc., in which they appear can be utilized by a growing seedling. They differ in structure from the fibrous celluloses and are probably not cell-wall building material. They appear to be a form of reserve carbohydrates, which differ from the glucose-polysaccharides in being condensed in, or as a part of, the external structural material rather than in the internal storage organs. They are soluble in water and exhibit the properties of gums, and are often classified with the gums and described under the names "galactans," "mannosans," "pentosans," etc.
The normal celluloses, of which the fibers obtained from cotton, flax, hemp, etc., are typical examples, are widely distributed in plants and form the commercial sources for all textile fibers of vegetable origin. Ordinary cotton fiber contains 91 per cent of cellulose, about 7.5 per cent of water, 0.4 per cent of wax and fat, 0.55 per cent of pectose derivatives, and 0.25 per cent of mineral matter; or a total of only 1.2 per cent of non-cellulose solids. Filter paper is practically pure cellulose.
Pure cellulose is a white, hygroscopic substance, which is insoluble in water and in most other solvents. If heated with water under pressure to about 260° C., it dissolves completely without decomposition. If boiled with a strong solution of zinc chloride, or treated in the cold with zinc chloride and concentrated hydrochloric acid, or with an ammoniacal solution of copper hydroxide (Schweitzer's reagent), it dissolves to a clear solution from which it may be reprecipitated without chemical change by neutralizing or diluting the solution.
Cellulose has the formula (C6H12O5)n. When hydrolyzed under the influence of the enzyme cytase, it breaks down, first into cellobiose, an isomer of maltose, and then into glucose. It is, therefore, chemically like, but not identical with, starch; and structurally it is arranged in fibrous form instead of in granules. Under the action of fermentative enzymes, as when vegetable matter decays under stagnant water, in swamps, etc., cellulose breaks down into carbon dioxide and marsh gas, according to the equation
(C6H12O5)n + nH2O = 3nCO2+3nCH4.
Cellulose is acted upon by caustic alkalies in a variety of ways. When fused with a mixture of dry sodium and potassium hydroxides, it is decomposed into oxalic and acetic acids. When heated with a 10 to 15 per cent solution of caustic soda, cellulose fibers thicken and become translucent, thus resembling silk fibers. This process, known as "Mercerizing," is largely used for the production of commercial fabrics.
Acids also act on cellulose in a variety of ways. When heated with nitric acid (sp. gr. 1.25), it is converted into oxycellulose; while dilute sulfuric acid, under similar conditions, yields hydro-cellulose, a substance having the formula C12H22O11, which retains the fibrous structure of the original cellulose but which, when dry, may be rubbed up into a fine powder. Concentrated nitric acid, or better, a mixture of concentrated nitric and sulfuric acids, acts upon cellulose, converting it into various nitro-derivatives, several of which have great industrial value. The number of NO3 groups which unite with the cellulose molecule under these conditions depends upon the temperature, pressure, etc., employed during the nitration process; di-, tri-, tetra-, penta-, and hexanitrates are all known. Pyroxylin, or collodion, is a mixture of the tetra- and penta-nitrates, which is soluble in alcohol and is used in surgery, in photography, and in the manufacture of celluloid, which is a mixture of collodion and camphor. The hexanitrate, C12H14(NO3)6O4, is the violent explosive known as gun-cotton.
Gentler oxidizing agents, such as "bleaching powder," etc., have no effect upon cellulose, and hence are extensively used in the treatment of cotton and other vegetable fibers, in preparation for their use in the manufacture of textiles, paper, etc.
Cellulose is indigestible in the alimentary tract of animals, but the putrefactive bacteria which are generally present there ferment it, with the production of acids of the "fatty acid" series, carbon dioxide, methane, and hydrogen. Excessive fermentations of this kind are responsible for the distressing phenomenon known as "bloat."
The compound celluloses comprise the larger proportion of the material of the woody stems of plants. They consist of a base of true cellulose, which is either encrusted with or chemically combined with some non-cellulose constituent. Depending upon the nature of the non-cellulose component, the compound celluloses are divided into three main groups, known respectively as (1)[Pg 74] ligno-celluloses, (2) pecto-celluloses, and (3) adipo-, or cuto-celluloses. As the names indicate, the non-cellulose component in the first group is lignin; in the second, pectic substances; and in the third, fats or waxes.
Ligno-celluloses.—In the young plant cell, the cell-walls consist of practically pure cellulose; but as the plant grows older, this becomes permeated with lignin, or woody fiber, until in the stem of a tree, for example, the proportion of cellulose in the tissue is only 50 to 60 per cent. In the preparation of wood pulp for the manufacture of paper, the lignin materials are dissolved off by means of various chemical reagents, leaving the cellulose fibers in nearly pure form for use as paper. The lignin material generally consists of two types of substances, one of which contains a closed-ring nucleus of unknown composition and the other is probably a pentosan. These materials are so extremely difficult to hydrolyze that their composition has not yet been definitely determined.
Pecto-celluloses are found in various species of flowering plants; those which are present in the stems and roots being true pecto-celluloses, while those which are found in fruits and seeds contain mucilages rather than pectose derivatives, and are generally designated as "muco-celluloses." The exceedingly inert character of these compounds makes their study difficult and their functions uncertain.
The term cuto-celluloses is applied to the group of substances, including suberin and cutin, which constitute waterproof cell-walls. These were formerly supposed to consist of true cellulose impregnated with fatty or wax-like materials. Recent investigations seem to indicate, however, that there is really no cellulose nucleus in such walls as these, but that they are compound glyceryl esters resembling the true fats (see Chapter X) in composition. If this view should finally be established as a fact, this sub-group of supposed compound celluloses should be dropped from consideration as such.
PHYSIOLOGICAL USE OF CELLULOSES
There seems to be no question that the sole use of celluloses is to serve as structure-building materials. They are undoubtedly elaborated from the carbohydrates as the cell grows. In only[Pg 75] rare cases, however, is there any evidence that they can be reconverted into carbohydrates to serve as food material. Certain bacteria can make use of cellulose as food, and secrete an enzyme, cytase, which aids in the hydrolysis of cellulose to sugars for this purpose. But this enzyme seems rarely, if at all, to be present in the tissues of higher plants. It has been reported that some cellulose is hydrolyzed during the malting of barley, indicating that this might have some food use for the growing seedling; but this observation has not been confirmed and later investigations seem to throw doubt upon its accuracy.
Bacteria of decay also act upon cellulose materials, converting them chiefly into gaseous products; but this seems to be a provision of nature for the destruction of the cell-wall material of dead plants, rather than an arrangement for the constructive use of it as food for the bacterium. When fibrous plant residues decay in the soil, the cellulose compounds are first converted into a series of complex organic acids, known as "humins," which undoubtedly have a significant effect upon the chemical and physical properties of the soil, but these have little interest or significance in a study of the chemistry of plant growth.
REFERENCES
Abderhalden, E.—"Biochemisches Handlexikon, Band 2, Gummisubstanzen, Hemicellulosen, Pflanzenschleimen ..." 729 pages, Berlin, 1911; and "Band 8—1 Ergänzungsband (same title as Band 2)—," 507 pages, Berlin, 1914.
Schwalbe, C. G.—"Die Chemie der Cellulose," 665 pages, Berlin, 1911.
CHAPTER VI
GLUCOSIDES
Strictly speaking, the term glucoside should be applied only to such compounds as contain glucose as the characteristic basic group. But in common usage, it refers to any compound which, when hydrolyzed, yields a sugar as one of the products of the hydrolysis. In all the natural glucosides which occur in plant tissues, the other organic constituent, which is represented by the R in the formula for glucosides (R·C6H11O5, or R·(CHOH)5CHO) is some aromatic group, or closed-ring benzene derivative.[3] The different organic constituents of glucosides are of a great variety of types, such as phenols, alcohols, aldehydes, acids, oxyflavone derivatives, mustard oils, etc. It is noteworthy, however, that no nitrogenous groups of the protein type have been found combined with sugars in glucosides.
Some glucosides contain more than one saccharide group, possibly as di- or trisaccharides. Under proper conditions of hydrolysis, one or more of the saccharide groups can be removed from such compounds, resulting in glucosides of simpler structure.
Most of the common glucosides are derived from d-glucose. Some are known, however, which are derivatives of galactose or rhamnose; while in some cases the exact nature of the sugar which is present has not yet been determined.
FOOTNOTES:
The structural formula for benzene, C6H6,  is one which it is difficult and inconvenient to reproduce in type. On that account, it is customary to indicate this formula by a plane hexagon, thus
is one which it is difficult and inconvenient to reproduce in type. On that account, it is customary to indicate this formula by a plane hexagon, thus  .
.
It is understood, in all such cases, that the figure represents six carbon atoms arranged in a closed ring, with alternate double and single bonds, and with a hydrogen atom attached to each carbon. The printing of some other group as OH, CH3, adjacent to an angle of the hexagon means that this group replaces the H atom in the compound which is being illustrated.
HYDROLYSIS OF THE NATURAL GLUCOSIDES
All natural glucosides are hydrolyzed into a sugar and another organic residue by boiling with mineral acids; although they vary widely in the ease with which this hydrolysis is brought about.
In most cases, the glucoside is easily hydrolyzed by an enzyme which occurs in the same plant tissue, but in different cells than those which contain the glucoside. Injury to the tissues, germination processes, and perhaps other physiological activities of the cells, result in bringing the enzyme in contact with the glucoside and the hydrolysis of the latter takes place. A large number of such enzymes have been found in plants, many of which hydrolyze only a single glucoside. However, two enzymes, namely, the emulsin of almond kernels, and myrosin of black mustard seeds, each hydrolyze a considerable number of glucosides. In general, emulsin will aid in the hydrolysis of any glucoside which is a derivative of β-glucose, and myrosin will help to split up any sulfur-containing glucoside. Glucosides which are derivatives of rhamnose require a special enzyme, known as rhamnase, for their hydrolysis.
The following reactions for the hydrolysis of arbutin and of amygdalin are typical of this action, and will serve to illustrate the general structure of these compounds:
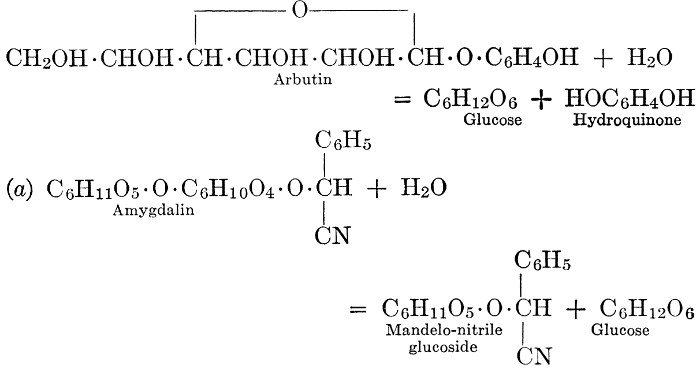
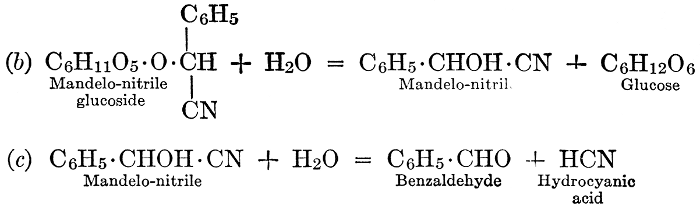
GENERAL PROPERTIES OF GLUCOSIDES
As a rule, glucosides are easily soluble in water. They are generally extracted from plant tissues by digestion with water or alcohol. In most cases, the enzyme which is present in other cells of the same tissue must be killed by heating the material, in a moist condition, to the temperature of boiling water, before the extraction is begun, as otherwise the glucoside will be hydrolyzed as rapidly as it is extracted from its parent cell. Maceration or otherwise bruising the tissue, after the enzyme has been destroyed, facilitates the extraction. The glucosides, after extraction and purification by recrystallization, are generally colorless, crystalline solids, having a bitter taste and levorotatory optical activity. This latter property is remarkable, as most of them are compounds of the strongly dextrorotatory d-glucose.
Many of the natural glucosides have marked therapeutic properties and are largely used as medicines; others are the mother-substances for brilliant dyes; for example, indican, from which indigo is obtained, and the alizarin glucosides.
Several hundred different glucosides have been isolated from plant tissues, and their properties described, and this number is being added to constantly, as the methods of isolation and study are improved. They may be classified into groups, according to the nature of the organic compound other than sugars which they yield when hydrolyzed. The following descriptions of the occurrence, constitution, products of hydrolysis, and special properties of typical members of each of the several different classes of glucosides will serve to illustrate their general relationship to plant growth.[Pg 79]
THE PHENOL GLUCOSIDES
Arbutin, C12H16O7, is obtained from the leaves of the bear berry (Arctostaphylos uva-ursi), a small evergreen shrub. When hydrolyzed by mineral acids or emulsin, it yields glucose and hydroquinone.
C12H16O7 + H2O = C6H12O6 + C6H4(OH)2.
Hydroquinone has strongly antiseptic properties. Arbutin is both an antiseptic and a diuretic, and is used in medicine.
Phloridzin, C21H24O10, is found in the bark of apple, pear, cherry, plum, and similar trees. Mineral acids (but not emulsin) hydrolyze it to glucose and phloretin (C15H14O5), according to the equation
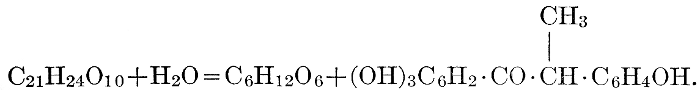
It is used in medicine as a remedy for malaria, having marked anti-periodic properties.
Glycyphyllin, C21H24O9, found in leaves of Smilax, yields rhamnose and phloretin, when hydrolyzed.
Iridin, C24H26O13 (glucose and irigenin), found in rootstocks of Iris, is used in medicine as a cathartic and diuretic.
Baptisin, C26H32O14·9H2O (two rhamnose and baptigenin), found in roots of wild indigo (Baptisia), has strong purgative properties.
Hesperidin, C50H60O27 (one rhamnose + two glucose + hesperitin), is found in the pulp of lemons and oranges.
The characteristic phenol group which is present in these glucosides has the following structural formula, in each case, the X indicating the H atom which is replaced by the sugar molecule to form the glucoside:
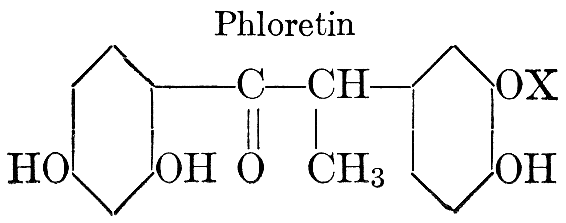
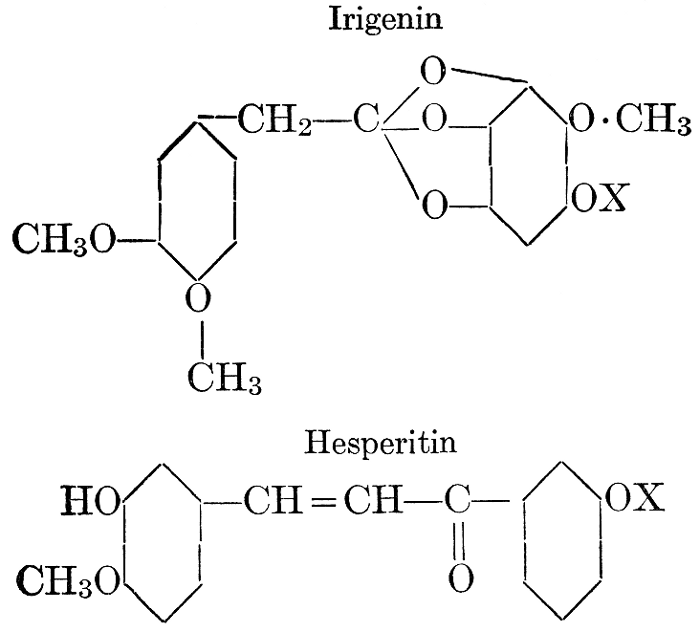
THE ALCOHOL GLUCOSIDES
Salicin, C13H18O7 (glucose + saligenin, or o-oxy benzyl alcohol) is found in the bark, leaves, and flowers of most species of willow, the proportion present depending upon the season of the year, and the sex of the tree. It is used as a remedy against fevers and rheumatism, causing less digestive disturbances than the salicylic acid which is the oxidation product of saligenin and which is sometimes used as a remedy for rheumatism.
Coniferin, C16H22O8 (glucose and coniferyl alcohol), is found in the bark of fir trees. The coniferyl alcohol obtained from coniferin by hydrolysis can be easily oxidized to vanillin, and is, therefore, the source for the artificial flavoring extract used as a substitute for the true extract of the vanilla bean.
Populin, C20H22O8 (glucose + saligenin+benzoic acid), found in the bark of poplar trees, is used in medicine as an antipyretic. It can be hydrolyzed, by a special enzyme, into salicin and benzoic acid.
The structure of the two typical closed-ring alcohols which are present in these glucosides is indicated by the following formulas;
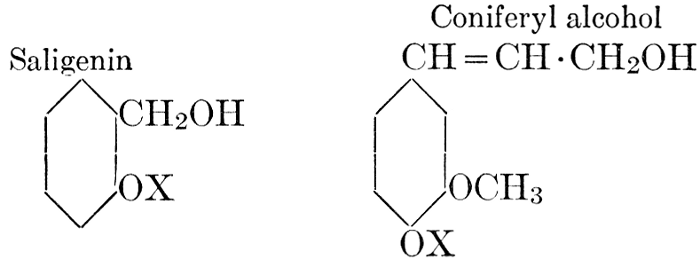
THE ALDEHYDE GLUCOSIDES
Salinigrin, C13H16O7 (glucose and m-oxy benzaldehyde), is found in the bark of one species of willow (Salix discolor). Its isomer, known as helicin (glucose and o-oxy benzaldehyde, or salicylic aldehyde), does not occur naturally in any plant, but is easily produced artificially by the gentle oxidation of salicin. Their relationships are shown on the following formulas;
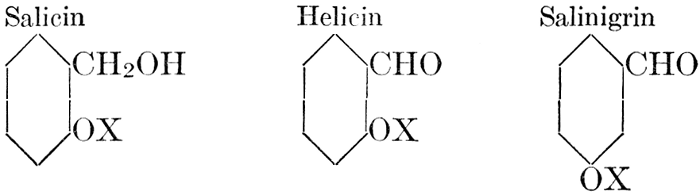
Amygdalin, also contains a benzaldehyde group, but there is linked with it a hydrocyanic acid group; hence, this glucoside is usually classed with the cyanophoric glucosides (see page 86).
THE ACID GLUCOSIDES
The most common example of this group is gaultherin, C14H18O8, which is found in the bark of the black birch and is a combination of glucose with methyl salicylate. Both the glucoside itself and the methyl salicylate ("oil of wintergreen") which is derived from it are used as remedies for rheumatism.
Jalapin, C44H56O16 (glucose and jalapinic acid), and convolvulin, C54H96O27 (glucose + rhodeose + convolvulinic acid), are glucosides of very complex organic acids, found in jalap resin, which are used in medicine as cathartics or purgatives.
THE OXY-CUMARIN GLUCOSIDES
Cumarin itself is widely distributed in plants. No glucoside containing cumarin as such has yet been isolated; but several glucosides of its oxy-derivatives are known. The following are common ones:
Skimmin, C15H16O8 (glucose and skimmetin), is found in Skimmia japonica; æsculin, C15H16O9 (glucose and æsculetin), is found in the bark of the horse-chestnut, Æsculus hippocastanum, and its isomer, daphnin (glucose and daphnetin), in several species[Pg 82] of Daphne; and fraxin, C16H18O10 (glucose and fraxetin), is found in the bark of several species of ash.
The structural arrangement of the oxy-cumarin groups which are found in these glucosides is shown in the following formulas. It is not known to which OH group the sugar is attached, in each case.
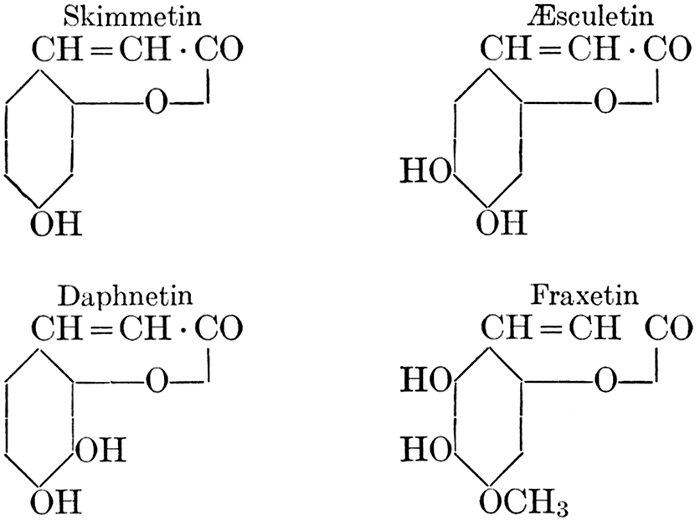
Scopolin, C22H28O14, found in Scopolia japonica, contains two glucose molecules united to a monomethyl ether of æsculin; while limettin, found in certain citrus trees, is the dimethyl ether of æsculin.
THE PIGMENT GLUCOSIDES
Many, if not all, of the red, yellow, violet, and blue pigments of plants either exist as, or are derived from, glucosides. These are of three types: the madder, or alizarin, reds are derivatives of various oxy-anthraquinones; most of the soluble yellow pigments are glucosides derived from flavones or xanthones; and the soluble red, blue, and violet pigments of the cell-sap of plants are mostly anthocyan derivatives. The four basic groups, or nuclei, which are present in these different types of compounds are complex groups consisting essentially of two benzene rings linked together through a third ring in which there are either two oxygen atoms in the ring, or one oxygen in the ring and a second attached to the opposite carbon in the (C=O) arrangement, as shown by the following diagrammatic formulas:[Pg 83]
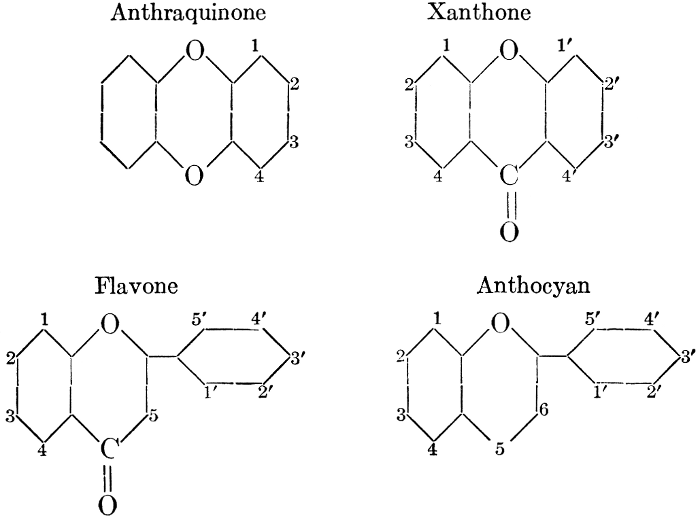
The red dyes which were formerly obtained from madder, the powdered roots of Rubia tinctoria, but are now almost wholly artificially synthetized, consist of at least four different glucosides, the organic group of which, in each case, is an hydroxy-derivative of anthraquinone. The most important of these is ruberythric acid, composed of two molecules of glucose linked with one of alizarin (1,2, dioxyanthraquinone). Xanthopurpurin contains 1,3, dioxyanthraquinone, which is isomeric with alizarin; and rubiadin is a monomethyl (the CH3 being in the 4 position), derivative of this compound. Purpurin is a glucoside of 1,2,4, trioxyanthraquinone.
The soluble yellow pigments are generally glucosides of hydroxy-derivatives of xanthone or flavone, known as oxyxanthones or oxyflavones. The sugars which are united to these nuclei vary greatly, so that there are a great variety of yellow, white, or colorless flavone or xanthone pigment compounds. These compounds are almost universally present in plants. For example, one typical set of examinations of the wood, bark, leaves, and flowers of over 240 different species of tropical plants showed that flavone derivatives were present in every sample which was tested, the pigments being usually located in the powdery coating of the epidermis of the tissues.
The following typical examples will serve to illustrate the composition and properties of the glucosides of this type.[Pg 84]
Quercitrin, C21H20O11, is found in oak bark, in the leaves of horse-chestnut, and in many other plants, often associated with other pigments. It is a brilliant yellow crystalline powder. Industrially, it ranks next to indigo and alizarin in importance as a natural dye stuff. It is a glucoside of rhamnose with 1,3,3',4', tetraoxyflavonol (i.e., the flavone nucleus with five OH groups replacing the hydrogens in the 1, 3, 5, 3', and 4' positions). Quercetin, C15H10O7, which is the tetraoxyflavonol itself, without any sugar in combination with it, is found in the leaves of several species of tropical plants and in the bark of others. Isoquercitrin, C21H20O12, is derived from the same flavone, but contains glucose instead of rhamnose, as the sugar constituent of the glucoside.
Apiin, C26H20O9, the yellow glucoside found in the leaves of parsley, celery, etc., contains apiose (a pentose sugar of very unusual structure, represented by the formula,
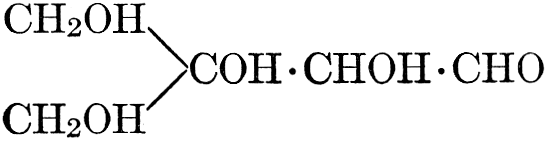
and apigenin, which is a 1,3,4', trioxyflavone.
Xanthorhamnin, C34H42O20, is a very complex glucoside containing two rhamnose and one galactose groups, united with rhamnetin, which is quercitin with the H of the OH in either the 1, or 3, position replaced by a methyl group. There are several similar pigments which differ from xanthorhamnin only in the number or position of the methoxy groups (i.e., the OH groups with a CH3 replacing the H), or in the nature of the sugar which is present in the compound. Rhamnetin itself is found in the fruits of certain species of Rhamnus, and is used in dyeing cotton.
The structural arrangement of the characteristic groups of these flavone pigments will be dealt with more in detail in the chapter dealing with Pigments (Chapter VIII).
The best-known yellow pigment which is a xanthone derivative is euxanthic acid, known as "Indian yellow," which is a "paired" compound of glucuronic acid (see page 42) and euxanthone. The latter is a 2, 3', dioxyxanthone. The pigment is found in the urine of cattle which have been fed on mango leaves.
The soluble red, blue, and violet pigments are glucosides of various hydroxy-derivatives of the anthocyan nucleus. Their constitution and properties will be discussed in detail in the chapter dealing with the Pigments. These compounds are iso[Pg 85]meric with similar flavone and xanthone derivatives, and the transition from one color to the other in plants takes place very easily under the action of oxidizing or reducing enzymes. This accounts for the change of reds and blues to yellows and browns, and vice versa, under changing temperature conditions.
The following red or blue plant pigments, which are anthocyan glucosides, have been isolated and studied (for the structural arrangement of the characteristic groups, see pages 116): from cornflower and roses, cyanin, C28H31O16Cl (2 molecules glucose + cyanidin); from cranberries, idain, C21H21O10Cl (galactose + cyanidin); from geranium, pelargonin, C27H30O15Cl (2 molecules glucose + pelargonidin); from pæony, pæonin, C28H33O16Cl (2 molecules glucose + pæonidin, a monomethyl cyanidin); from blue grapes, œnin, C23H25O12Cl (glucose + œnidin); from whortle berry, myrtillin, C22H23O12Cl (glucose + myrtillidin); from larkspur, delphinin, C41H39O21Cl (2 molecules glucose + 2 molecules p-oxybenzoic acid + delphinidin); and from mallow, malvin, C29H35O17Cl (2 molecules glucose + malvidin).
The blue dye, indigo, is derived from a glucoside of an entirely different type, known as indican. Indican is readily extracted from the leaves of various species of indigo plants. When hydrolyzed, it yields glucose and indoxyl (colorless). Indoxyl is easily oxidized to indigotin (the deep blue dye known as "indigo"). The equations illustrating these changes are as follows:
| (a) C14H17O6N | + | H2O | = | C6H12O6 | + | C8H7ON |
| Indican | Glucose | Indoxyl | ||||
| (b) 2C8H7ON | + | O2 | = | C16H10O2N | + | 2H2O |
| Indoxyl | Indigotin |
The structural relationships of indoxyl and indigotin may be illustrated by the following formulas:
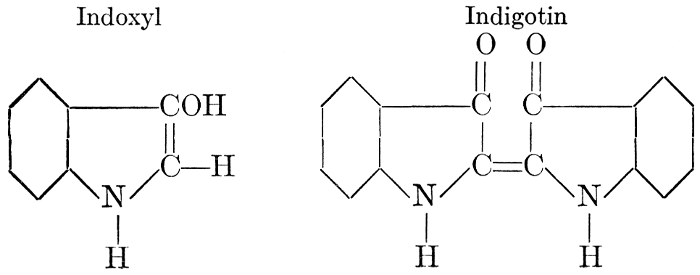
Natural indigo dye is prepared by fermentation of indigo leaves, the decay of the cell-walls liberating the enzymes in the tissues, which bring about the chemical changes illustrated in the above equations.
THE CYANOPHORE GLUCOSIDES
Several glucosides which yield hydrocyanic acid as one of the products of their hydrolysis are of common occurrence in plants. These are generally spoken of as the "cyanogenetic" glucosides; but as they do not actually produce cyanogen compounds, but only liberate them when hydrolyzed, the recently suggested term "cyanophore" undoubtedly more correctly indicates their properties.
The best known and most widely distributed of these is amygdalin. Amygdalin was first discovered in 1830, and was one of the first substances to be recognized as a glucoside. It is found in large quantities in bitter almonds and in the kernels of apricots, peaches, and plums; also in the seeds of apples, etc., in fact in practically all the seeds of plants of the Rose family. It is the mother substance for "oil of bitter almonds," which is widely used as a flavoring extract.
Amygdalin has been the object of very extensive studies, and even yet the exact nature of the linkage between its constituent groups is not certainly known. When completely hydrolyzed, it yields two molecules of glucose and one each of benzaldehyde and hydrocyanic acid. Recent studies indicate that the two sugar molecules are separately united to the other constituents, rather than united with each other in the disaccharide relationship. In other words, amygdalin is a true glucoside rather than a maltoside. This is indicated by the fact that when submitted to the action of all known hydrolyzing agents which affect it, it has never been found to yield maltose as one of the products of hydrolysis. Furthermore, the rate of hydrolysis of amygdalin is not affected by the presence of maltose; and the segregation of the two glucose molecules is accomplished by enzymes other than maltase, which is the only enzyme which is known to break up a maltose molecule. Since the exact nature of the linkage is not known, it is customary and convenient to indicate the unit groups as linked together in the following order:
| C6H11O5— | O—C6H10O4— | O—C6H5·CH | —C | ≡N |
| (1) | (2) | (3) | (4) |
A study of the hydrolysis reactions of amygdalin shows that there are three different linkages in the molecule which may be broken by the simple interpolation of a single molecule of water and a fourth which may be split by a different type of hydrolysis, namely, the C≡N linkage. These are indicated by the numbers below the corresponding portion of the formula above. Most hydrolyzing agents break the molecule first at (1), yielding one molecule of glucose and one of mandelo nitrile glucoside (see page 77). The next step usually breaks the latter at the point indicated by (2), yielding glucose and benzaldehyde cyanhydrin, or mandelo nitrile. The latter in turn breaks down at (3) into benzaldehyde and HCN. But when amygdalin is boiled with concentrated hydrochloric acid, the first change is the splitting off at (4) of the nitrogen in the form of ammonia and the consequent conversion of the CN group into a COOH group, producing amygdalinic acid. On further hydrolysis, this breaks up in the same order as before. Similarly, it is possible to convert mandelo nitrile into mandelic acid by splitting off the nitrogen to form a COOH group, instead of splitting off the HCN group leaving benzaldehyde.
The mandelo nitrile glucoside contains an asymmetric carbon atom which is wholly outside its glucose group, thus C6H10O5—O—C6H5·CH·CN. Hence, it may exist in dextro, levo, and racemic forms. In the amygdalin molecule, it exists in the dextro form, which has been named "prunasin." The levo form, known as "sambunigrin," has been obtained by hydrolysis of a compound isomeric with amygdalin, whose composition has not been definitely worked out; while the racemic form, known as "prulaurasin," has been prepared from isoamygdalin, by the action of alkalies. Hence, all the possible compounds indicated by the presence of the asymmetric carbon have been found and identified.
The crude enzyme preparation which is obtained from almond seeds, known as "emulsin," contains two enzymes, amygdalase, which breaks the amygdalin molecule at linkage (1), and prunase, which breaks it at (2). The action of amygdalase must always precede that of prunase. In other words, it is never possible to break off a disaccharide sugar from the molecule, either by the action of prunase alone, or by means of any other hydrolytic agent.
Dhurrin, C14H17O7N, is another glucoside of fairly general occurrence in plants, which yields HCN as one of the products of[Pg 88] its hydrolysis. It is found in the leaves and stems of several species of millets and sorghums. Frequent cases of poisoning of cattle from eating of these plants as forage have been reported. On hydrolysis, dhurrin first yields glucose and paraoxy-mandelo nitrile; the latter then breaks down into paraoxy-benzaldehyde and HCN.
Vicianin, C19H25O10N, is a cyanophoric glucoside, found in the seeds of wild vetch, etc. On hydrolysis, it yields glucose, arabinose, and d-mandelo nitrile. It is, therefore, similar to amygdalin, except that one glucose molecule is replaced by arabinose.
THE MUSTARD OIL GLUCOSIDES
The seeds of several species of plants of the Cruciferæ or mustard family contain glucosides in which the other characteristic group is a sulfur-containing compound. These glucosides yield "mustard oils" when they are hydrolyzed by the enzyme myrosin, which accompanies them in the plant. The following glucosides, found in the seeds of white and black mustard, are the best-known representatives of this class.
Sinigrin, C10H16O9NS2K, found in black mustard seeds, when hydrolyzed yields glucose, acid potassium sulfate, and allyl isosulfocyanide (mustard oil), as indicated by the equation.
C10H16O9NS2K + H2O = C6H12O6 + C3H5≡N=C=S+KHSO4.
The acid potassium sulfate group separates first and most readily, leaving a compound known as merosinigrin, for which the following formula has been suggested:
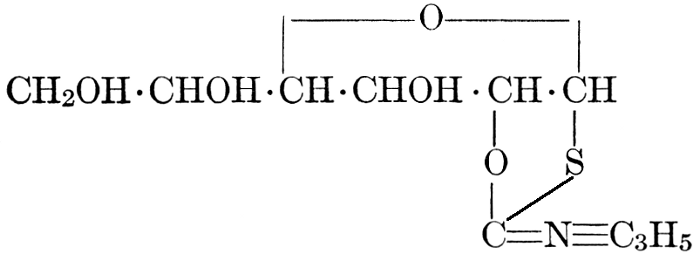
This compound usually breaks down into glucose and mustard oil; but by special treatment it is possible to obtain from it thioglucose, C6H11O5·SH. This indicates that in the original glucoside the glucose is linked with the mustard oil through the sulfur atom.[Pg 89]
Sinalbin, C30H42O15N2S2, from white mustard seeds, when hydrolyzed by myrosin, yields glucose, sinalbin mustard oil (a paraoxybenzyl derivative of allyl isosulfocyanide) and sinapin acid sulfate; according to the equation
| C30H42O15N2S2 | + | H2O | = | C6H12O6 | + | C7H7O·NCS | + | C16H24O5N·HSO4. |
| Sinalbin | Glucose | Sinalbin mustard oil | Sinapin acid sulfate |
The sinalbin mustard oil may be represented by the formula 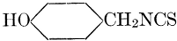 . Hydrolysis of the sinapin acid sulfate converts it into sinapinic acid, C6H2OH·(OCH3)2·CH=CH·COOH, choline, N(CH3)4C2H4OH (see page 152), and H2SO4. It is, therefore, a very complex glucoside.
. Hydrolysis of the sinapin acid sulfate converts it into sinapinic acid, C6H2OH·(OCH3)2·CH=CH·COOH, choline, N(CH3)4C2H4OH (see page 152), and H2SO4. It is, therefore, a very complex glucoside.
TEE DIGITALIS GLUCOSIDES
The five, or more, glucosides which are present in the leaves and seeds of the foxglove (Digitalis purpurea) have been extensively studied, as they are the active principles in the various digitalis extracts which are used in medicine as a heart stimulant.
Digitoxin, C34H54O11, which is the most active of these glucosides in its physiological effects, when hydrolyzed, yields digitoxigenin, C22H32O4, and a sugar having the formula C6H12O4, which is known as "digitoxose" and is supposed to be a dimethyl tetrose.
Digitalin, C35H56O14, is also strongly active. When hydrolyzed, it yields digitaligenin, C22H10O3, glucose, and digitoxose.
Digitonin, C54H92O28, constitutes about one-half of the total glucosides in the extract which is obtained from most species of the digitalis plants. It is much less active than the others. It is a saponin (see page 90) in type. On hydrolysis, it yields 2 molecules of glucose, 2 of galactose, and one of digitogenin.
Gitonin, C49H80O23, containing 3 molecules of galactose, one of a pentose sugar, and one of gitogenin; and gitalin, C28H48O10, containing digitoxose and gitaligenin, have also been isolated from digitalis extracts.
The structural arrangement of the characteristic groups in these glucosides has not yet been definitely worked out.[Pg 90]
Cymarin, the active principle of Indian hemp (Apocynum cannabinum), is similar in type to the digitalis glucosides. When hydrolyzed, it yields a sugar known as "cymarose," C7H14O7, which seems to be a monomethyl derivative of digitoxose, and cymarigenin, C23H30O5, a compound which is either identical or isomeric with the organic residue obtained from other members of this group.
THE SAPONINS
The saponins constitute a group of glucosides which are widely distributed in plants, whose properties have been known since early Grecian times. They have been found in over four hundred different species of plants, belonging to more than forty different orders.
The most characteristic property of saponins is that they form colloidal solutions in water which produce a soapy foam when agitated, and are peculiarly toxic, especially to frogs and fishes. In dry form, they have a very bitter, acrid taste, and their dust is very irritating to the mucous membranes of the eye, nose, and throat.
On hydrolysis, the saponins yield a variety of sugars,—glucose, galactose, arabinose, and sometimes fructose, and even other pentoses—and a group of physiologically active substances, known as "sapogenins."
The more toxic forms of these glucosides are known as "sapotoxins."
The chemical composition of the saponins varies so widely that it is scarcely possible to cite typical individuals. Sarsaparilla, the dried root of smilax plants, contains a mixture of non-poisonous saponins, from which at least four individual glucosides have been isolated and studied. Corn cockle contains a highly poisonous sapotoxin which, on hydrolysis, yields four molecules of a sugar and one of sapogenin, C10H16O2. Other sapotoxins are obtained from the roots of soapwort and from several species of Gypsophila. Digitonin and digito-saponin are glucosides of this type which are found in the extracts from various species of Digitalis.[Pg 91]
THE PHYSIOLOGICAL USES OF GLUCOSIDES
It is scarcely conceivable that substances which vary so widely in composition as do the different types of glucosides can possibly all have similar physiological uses in plants. The cyanophoric glucosides, the pigment glucosides, the mustard oil glucosides, and the saponins, for example, can hardly be assumed to have the same definite relationships to the metabolism and growth of the plant. To be sure, they are alike in that they all contain one or more sugar molecules, and it is probable that the carbohydrates which are held in this form may serve as reserve food material, especially when the glucoside is stored in the seeds; but it is obvious that the simpler and more normal form of such stored food is that of the polysaccharides which contain no other groups than those of the carbohydrates. It seems much more probable that the physiological uses of glucosides depend upon their ability to form temporarily inactive "pairs" with a great variety of different types of organic compounds which are elaborated by plants for a variety of purposes.
It has been noted that in most, if not all, instances, the glucosides are accompanied in the same plant tissue (although in separate cells) by the appropriate enzyme to bring about their hydrolysis and so set free both the sugar and the other characteristic component whenever the conditions are such as to permit the enzyme to come in contact with the glucoside. This occurs whenever the tissue is injured by wound or disease, and also during the germination process.
Injury to the plant tissue seems to be a necessary preliminary to the functioning of the active components of the glucoside, except in the case of the seeds. This leads naturally to the supposition that at least some of these glucosides are protective or curative agents in the plant tissues. This conception is further supported by the facts that many of the non-sugar components of glucosides are bactericidal in character and that the glucosides commonly occur in parts of the plant organism which are otherwise best suited to serve as media for the growth of bacteria. Thus, it is known that in the almond, as soon as the tissue is punctured, amygdalin is hydrolyzed and all bacterial action is inhibited. Similarly, the almost universal presence of glucosides containing bactericidal constituents in the bark of trees insures[Pg 92] natural antiseptic conditions for all wounds of the outer surfaces of the stem of the plant. In fact, it is easily conceivable that at least one of the reasons for the failure of the processes of decay of plant tissues to set in until after the death of the cells, is that during living, respiratory activity these antiseptic glucosides are so generally present in the tissues.
Further, it has been fairly well established that the "chromogens," or mother-substances of the pigments, which, under the influence of oxidase enzymes, serve to regulate the respiratory activities of the plant are essentially glucosidic in character. This, and other, functions of the pigments, most of which are glucosides, will be discussed at some length in the chapter dealing with the Pigments (Chapter VIII).
Many gaseous anæsthetics are known to have a marked effect in stimulating plant growth. In a number of cases, it has been shown that the contact of plant tissues with these anæsthetics brings about an interaction of the enzyme and glucoside which are present in the tissue, with the consequent hydrolysis of the latter, setting free its characteristic components. This observation has led to the supposition that many of the organic constituents of glucosides are definite plant stimulants, to which the name "hormones" has been applied. There is considerable experimental evidence to support this conception that glucosides may be the source of stimulating hormone substances, which will be discussed more in detail in the chapter dealing with these plant stimulants (Chapter XVII).
Glucosides may also serve as the mechanism for putting out of action of harmful products which may appear in the tissues as the result of abnormal conditions. These harmful substances may be rendered soluble by combination with sugars and so transposed by osmosis to some other part of the plant. The abnormally large percentages of glucosides which are present in certain species of plants during unfavorable climatic conditions lends some support to this view.
Finally, it may be assumed that easily oxidizable substances, such as aldehydes and acids, are possibly protected against too rapid, or premature, oxidation by being transformed into glucosides.
In general, it may be said that the glucosides seem to serve as the regulatory, protective, and sanatory agencies of the plant mechanism.[Pg 93]
BIOLOGICAL SIGNIFICANCE OF GLUCOSIDES
The bitter taste of glucosides and their almost universal presence in the bark of plants undoubtedly helps to prevent the destructive gnawing of the bark by animals.
Glucosides having either a strong bitter taste, or pronouncedly poisonous properties, likewise undoubtedly serve to protect such important organs of plants as the seeds and fruits from being prematurely eaten by birds and animals. The common disappearance of these bitter substances as the seed or fruit ripens adds to the attractiveness of the material for food for animals at the proper stage of ripeness to provide for wider distribution of the seeds for further propagation. Further, the very general occurrence of these protective glucosides in many of the vegetative parts of plants during the early stages of growth, followed by their disappearance after the seeds of the plant have been formed, certainly serves to protect these plants from consumption as forage by animals before they have been able to develop their reproductive bodies. The lack of palatability, and even the production of digestive disorders resulting from the eating of unripe fruit may be due, in part at least, to the presence of protective glucosides in unripe fruits and vegetables.
On the other hand, the almost universal presence of the brilliant pigment glucosides in the external parts of flowers undoubtedly serves to attract the insects which are biologically adapted to provide for the transportation of pollen from one blossom to another and so to insure the cross-fertilization which is so important in maintaining the vigor of many species of plants.
It is apparent that this important group of compounds, with its exceedingly varied and complex constituent groups, may play a variety of significant rôles in plant growth.
References.
Armstrong, E. F.—"The Simple Carbohydrates and Glucosides," 239 pages, Monographs on Biochemistry, London, 1919 (3d ed.).
Van Rijn, J. J. L.—"Die Glykoside," 511 pages, Berlin, 1900.[Pg 94]
CHAPTER VII
TANNINS
Using the term in its general application to a group of substances having similar chemical and physical properties, rather than in its limited application to a single definite chemical compound known commercially as "tannin," the tannins are a special group of plant substances, mostly glucosides, which have the following characteristic properties. First, they are non-crystalline[4] substances, which form colloidal solutions with water, which have an acid reaction and a sharp astringent taste. Second, they form insoluble compounds with gelatine-containing tissues, as shown by the conversion of hide into leather. Third, they form soluble, dark-blue or greenish-black compounds with ferric salts, the common inks. Fourth, they are precipitated from their solutions by many metallic salts, such as lead acetate, stannous chloride, potassium bichromate, etc. Fifth, they precipitate out of solution albumins, alkaloids, and basic organic coloring matters. Finally, most tannins, in alkaline solutions, absorb oxygen from the air and become dark brown or black in color.
FOOTNOTES:
[4] The needle-like forms, in which commercial "tannin" comes on the market, are not true crystals, but are broken fragments of the threads into which the colloidal tannin is "spun-out" from the syrupy extracts of nutgalls, etc.
OCCURRENCE
Tannins occur widely distributed in plants. Practically every group of plants, from the fungi up to the flowering plants, contains many species of plants which show tannin in some of their tissues. Among the higher plants, tannins occur in a great variety of organs. Thus, they are found in the roots of several species of tropical plants; in the sterns, both bark and wood, of oaks, pines, hemlock, etc.; in the leaves of sumac, rhododendron, etc.; in many fruits, especially in the green, or immature, stages; and in[Pg 95] the seeds of several species, either before or after germination. Tannins are also found in certain special structures, such as gland cells, cells of the pulvini, laticiferous tissues, etc. Further, they are especially abundant in the pathological growths known as galls, which often contain from 40 to 75 per cent of tannin and constitute the most important commercial source for these materials.
The principal commercial sources of tannin, which is used in the manufacture of inks, in the tanning of leather, in certain dyeing operations, etc., are oak-galls, the bark and wood of oak, hemlock, acacia, and eucalyptus, the bark of the mangrove, the roots of canaigre, and the leaves of several species of sumac.
CHEMICAL CONSTITUTION
Tannins are either free phenol-acids or, more commonly, glucosides of these acids. Common "tannin," when hydrolyzed, yields from 7 to 8 per cent of glucose, which indicates that it is a penta-acid ester of glucose, i.e., each glucose molecule has five acid groups attached to it. The formula for such a tannin is, therefore, as follows,
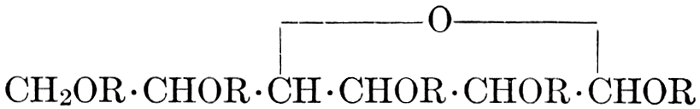
in which the R represents a complex phenol-acid like tannic acid, or digallic acid. These acids are derivatives of the common phenols, whose constitution will be brought to mind by the following series of formulas:
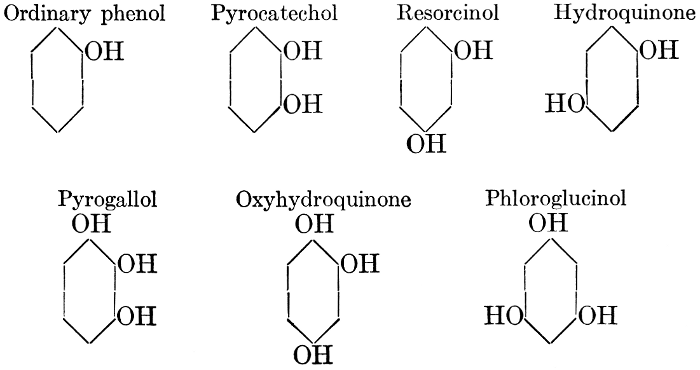
These phenols themselves do not occur as constituents of tannins, although they are often found in other glucosides, gums, etc. The following mono-carboxyl acid derivatives of these phenols are, however, found both free and in glucoside formation as constituents of many of the common tannins.
Pyrocatechuic acid, derived from pyrocatechol, represented by the formula,
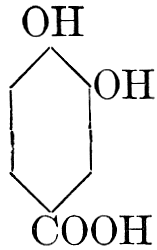
Gallic acid, derived from pyrogallol, and represented by the formula,
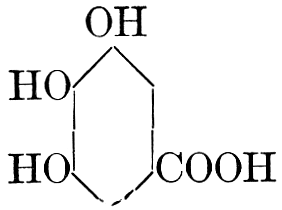
In most of the common tannins, however, the characteristic acids are oxy-derivatives of the so-called "tannon" group, represented by the formula, C6H5·CO·O·C6H5. For example, digallic acid, which is a constituent of many common tannins, is a tetra-oxy, mono-carboxyl derivative of this group, having the structural formula,
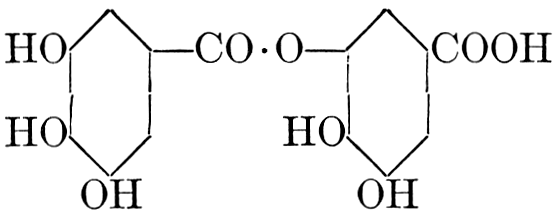
Ellagic acid, which is an hydrolysis product of many of the pyrogallol tannins (see below) and which produces the characteristic "bloom" on leather tanned by this type of tannins, has the following formula,
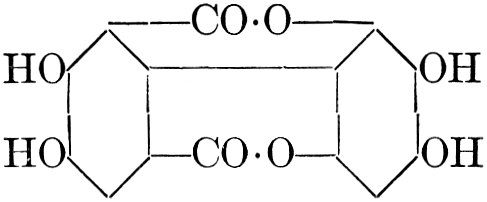
CLASSES OF TANNINS
The tannins are divided into two general classes, known respectively as the pyrogallol tannins and the catechol tannins. These differ in their characteristic reactions as follows:
| Pyrogallol variety | Catechol variety | |
|---|---|---|
| Ferric salts | Dark blue | Greenish black |
| Bromine water | No precipitate | Yellow or brown precipitate |
| Leather | Produce a "bloom" | No "bloom" |
| Conc. sulfuric acid | Yellow or brown | Red or pink |
| Lime water | Gray or blue ppte. | Pink to brown ppte. |
Pyrogallol tannins contain approximately 52 per cent of carbon; while the catechol tannins usually contain 59 per cent to 60 per cent, the difference being due to the absence of glucose from the molecule in the latter types.
The two types are distributed in plants as follows: pyrogallol tannins in oak-galls, oak wood, sumac, chestnut, divi-divi, and algaro billa; catechol tannins in the barks of pines, hemlocks, oaks, acacias, mimosas, cassia, and mangrove, in quebracho wood, canaigre roots, cutch and gambier. The so-called "pseudo-tannins" (i.e., compounds which do not tan leather but possess other properties like tannins) are found in hops, tea, wine, fruits, etc.[Pg 97]
SOME COMMON TANNINS
Ordinary commercial "tannin," or "tannic acid," is a compound of one molecule of glucose with five of digallic acid. It is found in many plants, and is prepared commercially from the Turkish oak-galls and the Chinese sumac-galls. It exhibits all the characteristic properties which have been listed above for tannins in general and responds to all the characteristic reactions of a pyrogallol tannin. It is extensively used for the manufacture of blue-black ink, and in many technical processes.
Catechu tannin and catechin are compounds of the catechol tannin type. The latter is obtained from acacia wood, mahogany wood, mimosa wood, etc. It is not a true tannin, since it does not convert hide into leather; but when heated to 120° or above, it is easily dehydrated, forming catechu tannin which is identical with that which is obtained directly from gambier and Bombay cutch (products made by evaporating water extracts from the bark of[Pg 98] various tropical trees). This latter is a true tannin, which is much used in dyeing and other technical processes.
"Quercitannic acid," obtained from oak bark, etc., is likewise a catechol tannin. It yields no glucose on hydrolysis.
A great many other tannins are known, and their possibilities for technical use in tanning, dyeing, etc., have generally been investigated; but so little has been learned about their composition and relation to the plant's own needs, that it seems unnecessary to discuss them in detail here.
PHYSIOLOGICAL USES OF TANNINS
Tannins are probably not direct products of photosynthesis. They are, however, elaborated in the green leaves of plants and translocated from there to the stems, roots, etc. Their close association with the photosynthetic carbohydrates has led many investigators to seek to establish for them some significant function as food materials, or as plastic substances in cell metabolism. Many conflicting views have been advanced, but a careful review of these leads inevitably to the conclusion that tannins probably do not serve in any significant way as food material. The glucose which is generally present in the tannin molecule may, of course, serve as reserve food material, but it seems probable that it functions as a constituent of the tannins only to assist in making them more soluble and hence more easily translocated through the plant tissues.
Some fungi, and perhaps other plants as well, can actually utilize tannins as food material under suitable conditions and in the absence of a proper supply of carbohydrates. But this does not prove that tannins can normally replace carbohydrates as food material for these species of plants.
There seems to be ample evidence that tannins are elaborated where intense metabolism is in progress, such as occurs in green leaves during the early growing season; in the rapid tissue formation which takes place after the stings of certain insects, producing galls, etc.; during germination, and as a result of any other unusual stimulation of metabolism. It may be, therefore, that tannins serve as safety accumulations of excessive condensations of formaldehyde, or other photosynthetic products, under such conditions. It seems certain that in all such cases tannins are the result of,[Pg 99] and not (as some investigators have supposed) the causative agents for, the abnormally rapid metabolism.
It seems to be fairly well demonstrated that tannins are intermediate products for the formation of cork tissue. This may account for their common occurrence in the wood and bark of trees. Indeed, it has been shown that gallic and tannic acids are present in considerable proportions in those parts of the plant where cork is being formed. Further, that they bear direct relation to cork-formation has been demonstrated in two different ways. First, cork-like substances have been artificially produced by passing a stream of carbon dioxide through mixtures of formaldehyde with various tannic acids. Second, by various treatments of cork, decomposition compounds showing tannin-like properties may be obtained.
Some investigators have held that not only cork tissue but also other lignose, or cell-wall material, may be developed from tannins. Certain observations with Spirogyra seem to indicate that tannin may play an important part in the formation of new cell walls during conjugation, as cells which are ready to conjugate are rich in tannin, which gradually diminishes in quantity until it is practically absent at the time of spore-formation. There seems to be no evidence that tannins perform any such function as this in higher plants, however.
Again, tannins may play a very important part in pigment-formation. They are very similar in structure to the anthocyanin pigments, both being made up of practically identical decomposition units, the phenolic bodies. The disappearance of tannins during the process of ripening of fruits may be connected, in part at least, with the development of the brilliant red, blue, and yellow pigments which give such rich colors to the thoroughly ripe fruits.
Finally, certain of the tannins undoubtedly serve as protective agents to prevent the growth of parasitic fungi in fruits, etc. Recent investigations show that at least some of the varieties of fruits which are resistant to the attacks of certain parasitic diseases utilize tannins for this purpose. This protective effect may be accomplished in two different ways. Either the tannin actually serves as an antiseptic to prevent the growth of the parasitic fungus within the tissues of the host plant, or it assists in the development of a corky layer which "walls-off" the infected area and so prevents further spread of the disease through the tissue.[Pg 100] Examples of both types of protective action have recently been reported.
It is obvious that the different forms of tannins may play different rôles in plant life, and the same tannin substance may possibly serve different purposes under different conditions.
BIOLOGICAL SIGNIFICANCE OF TANNINS IN FRUITS
The presence of tannins in fruits and the changes which they undergo during the ripening process cannot fail to attract attention to their biological significance in serving to protect the fruit from premature consumption as food by animals.
Tannins are of frequent occurrence in green fruits, imparting to them their characteristic astringent taste. They nearly always disappear as the fruit ripens. The fact that during the ripening process both sugars and fruit esters, as well as attractive surface pigments, are developed has led certain investigators to the conclusion that tannins serve as mother-substances for these materials in the green fruits and are converted into these attractive agencies during ripening. There is nothing in the chemical composition of tannins which indicates, however, that they are precursors of sugars or fruit esters, although (as has been pointed out) they may give rise to anthocyan pigments.
Further, recent researches concerning the tannin of persimmons (the best-known and most striking example of the phenomena under discussion) clearly show that the tannin is not actually used up during the ripening process; that instead it remains in the ripe fruit in practically undiminished quantity; but that when the fruit is ripe, the tannin is enclosed in certain special large cells or sacs, which are surrounded by an insoluble membrane, so that when the fruit is eaten by animals the astringent tannin, enveloped in these insoluble sacs, passes by the organs of taste of the animal without causing any disagreeable effects. This walling-off of the astringent tannins can be stimulated in partially ripe fruits by treating them with several different chemical agents, the simplest method being that of placing the unripe fruit in an atmosphere of carbon dioxide gas for a short period. The artificial "processing" of persimmons to render them edible for a longer period before they become naturally fully ripe and subject to decay is now a commercial enterprise. This process is of interest because of[Pg 101] its possible connection with the conversion of tannins into cork, under the influence of carbon dioxide gas, as mentioned in a preceding paragraph.
From these facts, it is apparent that in persimmons, and probably in other tannin-containing fruits, the process of natural selection has developed a mechanism for the secretion of tannin in green fruits, followed by a process for walling it off in harmless condition when the fruit is ripe, which serves most admirably to protect the fruit from consumption by animals before the enclosed seeds have fully developed their reproductive powers.
References.
Abderhalden, E.—"Biochemisches Handlexikon, Band 7, Gerbstoffe, Flechtenstoffe, Saponine, Bitterstoffe, Terpene, Aetherische Oele, Harze, Kautschuk," 822 pages, Berlin, 1912.
Allen's Commercial Organic Analysis, Vol. 5, "Tannins, Dyes and Coloring Matters, Inks," 704 pages, 6 figs., Philadelphia, 1911 (4th ed.).
Cook, M. T. and Taubenhaus, J. J.—"The Toxicity of Tannin," Delaware College Agricultural Experiment Station Bulletin No. 91, 77 pages, 43 figs., Newark, Del., 1911.
Dekker, J.—"Die Gerbstoffe," 636 pages, 3 figs., Berlin, 1913.
Gore, H. C.—"Experiments on the Processing of Persimmons to Render them Nonastringent," U. S. Department of Agriculture, Bureau of Chemistry Bulletin No. 141, 31 pages, 3 plates, 1911; and No. 155, 20 pages, 1912.
Lloyd, F. E.—"The Tannin-Colloid Complexes in the Fruit of the Persimmon, Diospyros," in Biochemical Bulletin, Vol. 1, No. 1, pages 7 to 41, 34 figs., New York, 1911.[Pg 102]
CHAPTER VIII
PIGMENTS
Practically all plant structures contain pigments. These may be considered as of two types: (a) the vegetative pigments, which have a definite energy-absorbing rôle in the metabolic processes of the tissues which contain them, and (b) the ornamental pigments. It is probable that the same chemical compound may serve in either one of these capacities under different conditions, but, in general, it is possible to assign either a definite vegetative, or physiological, use, or else a simple ornamental, or biological, significance to each of the common pigments. The first type is found widely distributed through the protoplasm, or cell-sap, of the plant structures; while the ornamental pigments are located chiefly in the epidermal cells, especially of flowers.
With respect to their colors, the plant pigments may be grouped as follows:
Green—the chlorophylls.
Yellow—the carotinoids, flavones, and xanthones.
Red—phycoerythrin, lycopersicin, anthocyanin.
Blue—anthocyan derivatives.
Brown—phycophæin, fucoxanthin.
Of these, the chlorophylls, the carotinoids, phycoerythrin (in red sea-weeds) and phycophæin (in brown sea-weeds) are generally vegetative pigments; while the others form the basis for most of the ornamental pigments, although they may have a definite energy-absorbing effect, in some cases.
THE CHLOROPHYLLS
The importance of the green coloring matter in plants has been understood for more than a century, its connection with[Pg 103] photosynthesis having been known as far back as 1819. But definite knowledge as to its chemical constitution is of very recent origin. As recently as 1908, it was asserted that chlorophyll is a lecithin-like body, yielding choline and glycero-phosphoric acid on hydrolysis. It is now known, however, that chlorophyll contains neither choline nor phosphorus, the earlier observations being due to mixtures of various other materials with the true chlorophyll in the extracts which were examined. Beginning with 1912, Willstätter and his collaborators, in a series of classic papers which were finally collected in book form, clearly demonstrated the chemical constitution of the green pigments of plants, which had been previously designated under the single name "chlorophyll." In 1912, Willstätter and Isler first showed that the green coloring matter which is extracted from plants by alcohol, ether, etc., is made up of two definite chemical compounds, to which they assigned the names "chlorophyll a" and "chlorophyll b," associated with two yellow pigments, carotin and xanthophyll, and, in some cases, with the reddish-brown fucoxanthin. The percentages of total pigment materials, and the relative proportions of the five different pigments, in several types of plants, are as follows:
| Land Plants, Per Cent. |
Brown Seaweeds, Per Cent. |
Green Algæ, Per Cent. |
|
|---|---|---|---|
| Total pigment in the dry matter | 0.99 | 0.29 | 0.21 |
| Proportion of: | |||
| Chlorophyll a | 63 | 55 | 44 |
| Chlorophyll b | 22 | 4 | 31 |
| Carotin | 6 | 11 | 7 |
| Xanthophyll | 9 | 10 | 18 |
| Fucoxanthin | 20 |
The two chlorophylls have the following formulas: chlorophyll a, C55H72O5N4Mg, and chlorophyll b, C55H70O6N4Mg. Hence, they differ only in having two hydrogen atoms in the one replaced by one oxygen atom in the other. Both are amorphous powders, from which crystalline chlorophyll (see below) can be obtained by hydrolysis. Chlorophyll a is blue-black, is easily soluble in most organic solvents, and when saponified by alcoholic potash gives a[Pg 104] transient pure yellow color. Chlorophyll b is dark green, is somewhat less soluble than the other form, and when saponified by potash gives a transient brilliant red.
Amorphous and Crystalline Chlorophyll.—When the chlorophyll of plants is extracted by alcohol and the alcoholic extract evaporated nearly to dryness, beautiful dark green crystals are obtained. Willstätter has shown, however, that in these crystallized forms the ethyl group (from the ethyl alcohol used) has replaced the phytyl group (see below) which is present in the pigments as they exist in the plant tissues; and that, when extracted by other solvents than alcohol, the pigments may be obtained in the amorphous forms in which they exist in the plant.
This change from amorphous to crystalline compounds may be understood from the preliminary statement that the chlorophylls are esters of tri-basic acids, in which one acid hydrogen is replaced by the methyl (CH3) group and a second by the phytyl (C20H39, from phytol, or phytyl alcohol, C20H39OH) group. When treated with ethyl alcohol (C2H5OH) for the purpose of extracting the pigments, the ethyl (C2H5) group replaces the phytyl group, thus yielding a methyl-ethyl ester, and these esters are the crystalline forms of the chlorophylls. This replacement is made possible through the action on the original pigment in the tissues of an enzyme, chlorophyllase, which is also present in the tissues, which splits off the phytyl group, forming phytyl alcohol, and leaving a free COOH group in the pigment, with which the alcohol used in the extraction forms the ethyl ester (see Chapter IX for a discussion of the formation and hydrolysis of esters).
While the chlorophylls are tri-basic acids, only two of the acid COOH groups actually function in ester-formation. The third acid group seems not to exist as a free acid group; but in chlorophyll a, it is in what is known as the "lactam" arrangement, represented by the —CONH— group, and in chlorophyll b, it is probably in the "lactone" arrangement, represented by the —COO— group; the two bonds in each case being attached to different structural units in the molecule (see page 106).
The change from amorphous to crystalline forms may be represented by the following formulas, in which the R represents the whole of the complex group to which the acid ester groups are united:[Pg 105]
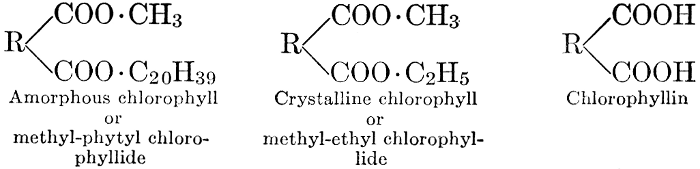
"Chlorophyllin," the compound in which the ester groups have been converted into free acid groups, as indicated above, may be obtained from either amorphous or crystalline chlorophyll by treatment with caustic potash dissolved in methyl alcohol.
Phytol.—This alcohol, which furnishes the characteristic ester group in the chlorophyll of plants, is a compound of very unusual composition, which has never been found in any other form or in any other type of compound which is present in either plant or animal tissues. Careful studies of its addition and oxidation products prove that it has the following structural arrangement:
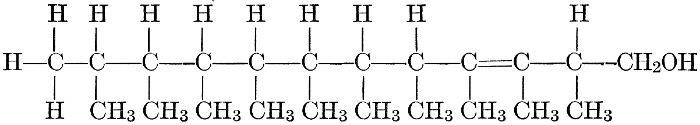
As this formula indicates, the compound contains one unsaturated, double-bond linkage, one primary alcohol group, and eleven methyl groups. As has been said, this alcohol occurs nowhere else in nature, and its presence and function in the chlorophyll molecule are, as yet, wholly unexplainable. Phytol itself is a colorless, oily liquid, with a high boiling point (145° in vacuo, 204° at 10 mm. pressure).
THE CONSTITUTION OF THE CHLOROPHYLLS
As has been mentioned, chlorophyll a differs from chlorophyll b by having one more oxygen and two less hydrogen atoms in the molecule, and in having one of its nitrogen atoms in the "lactam" arrangement. These differences in structure are represented by the following formulas which are commonly used to represent the two compounds, but which do not show the arrangements of the major groups of the complex molecules:[Pg 106]
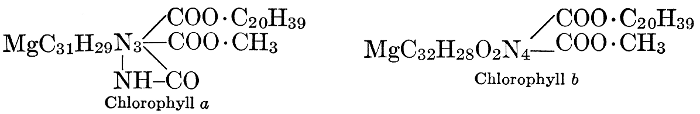
The chlorophylls are unstable compounds, readily acted upon by acids or alkalies, and by the enzyme chlorophyllase, which splits off the phytyl alcohol group. The progressive action of acids and of alkalies in breaking down the molecule, and the products of its oxidation and reduction, have served to establish the chemical composition of the compound in each case. Because of the importance of these pigments in the whole metabolic processes of the plant, it seems to be desirable to consider the nature of these reactions in some detail, as follows:
Decomposition of the Chlorophylls by Alkalies.—The first action of dilute alkalies on the chlorophylls is to split off, by hydrolysis, the alcoholic groups of the esters, producing the crystalline tri-basic acids, or chlorophyllins a and b. Each of these chlorophyllins exists in two forms, the normal and the iso, in which the attachment of the COOH groups to the other groups in the molecule is in different positions. Hence, chlorophyll a yields chlorophyllin a and isochlorophyllin a, and chlorophyll b yields chlorophyllin b and isochlorophyllin b, all four of which are tri-basic acids.
These compounds, when heated with alkalies, split off carbon dioxide in successive stages, losing one COOH group at each step, thus yielding a series of simpler compounds of the following types: First, di-basic acids; second, monobasic acids; and finally, ætiophyllin, a compound in which no COOH group is present. In all of these compounds, derived from chlorophylls by the action of alkalies, the Mg remains in the molecule, and all the Mg-containing derivatives from the chlorophylls are known as "phyllins." At the stage at which only one COOH group remains in the molecule, only one group arrangement is possible, and the derivatives from chlorophyllin a and isochlorophyllin b, and those from chlorophyllin b and isochlorophyllin a, are identical. At the final stage, the derivatives from all four forms are identical. This may be graphically illustrated by the following diagram indicating the progressive decomposition of the two chlorophylls under the action of alkalies:[Pg 107]
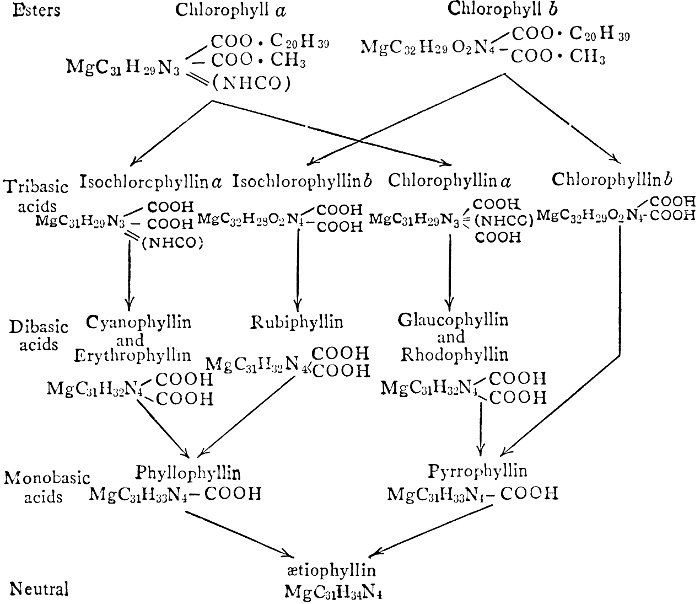
Decomposition of Chlorophylls by Acids.—The first action of dilute acids upon chlorophylls is to remove the magnesium, without otherwise changing the molecule. Two hydrogens go in in the place of the magnesium. Dilute acids act in precisely the same way upon each of the "phyllins" shown in the above scheme. In this way, a whole series of compounds, corresponding to each of the chlorophylls and their alkali-decomposition products, but with the magnesium lacking in each case, has been prepared. Thus,
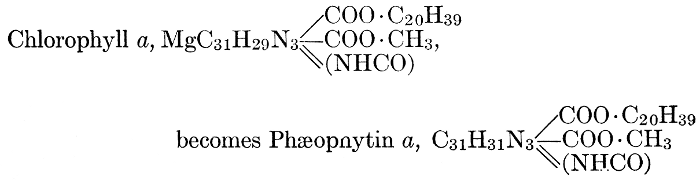
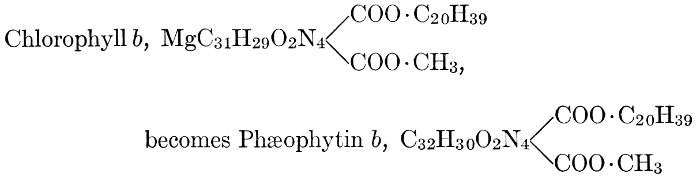
Similarly,
Isochlorophyllin a, becomes Phytochlorin e, Chlorophyllin a, becomes Phytochlorin f, and g,
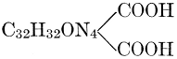
Isochlorophyllin b, becomes Phytorhodin g Chlorophyllin b, becomes Phytorhodin i and k,
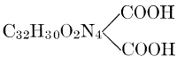
And bodies known as "porphyrins" are similarly derived from all the other known phyllins.
For example: cyanophyllin, MgC31H32N4(COOH)2, becomes cyanoporphyrin, C31H34N4(COOH)2; ætiophyllin, MgC31H34N4, becomes ætioporphyrin, C31H36N4, etc.
Phytochlorin e and phytorhodin g are the chief products of the decomposition by acids of the chlorophylls. Indeed, it was the production of these compounds which led to the discovery of the existence of the two chlorophylls. When treated with alkalies, they lose their carboxyl groups and become ætioporphyrin.
Decomposition of the Chlorophylls by Oxidation and Reduction.—When acted upon by oxidizing agents, such as chromic acid, the porphyrins yield two chief oxidation products, which are pyrrole derivatives having the following formulas,
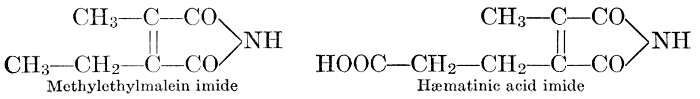
By reduction, there have been obtained from the chlorophylls and the various porphyrins, three isomeric pyrrole derivatives having the following formulas,
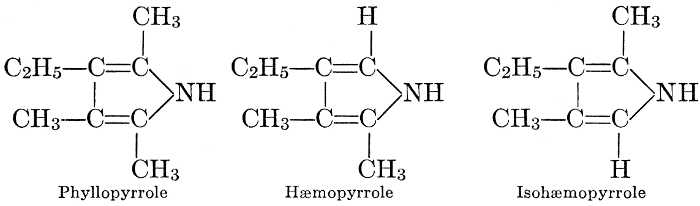
As a result of the study of these decomposition units, Willstätter has suggested the following formulas for the structural arrangement of ætiophyllin and ætioporphyrin, the compounds which result from the removal of all of the acid groups and finally of the magnesium from the chlorophylls,
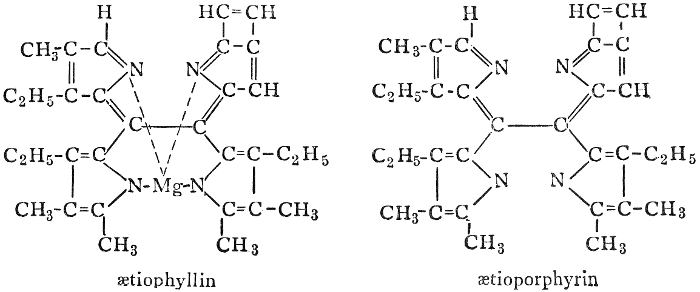
The COOH groups which are attached to these compounds to form the various phyllins and porphyrins, as well as the original chlorophylls, are supposed to be attached to the C2H5 groups in the above formulas, the different modifications, or compounds, depending upon the position in which one or more of these attachments are made.
SIMILARITY OF CHLOROPHYLL AND HÆMOGLOBIN
It seems to be desirable, at this point, to call attention to the remarkable similarity in the chemical composition of chlorophyll, the most important pigment of plants, and hæmoglobin, the all-important respiration-regulating pigment in the blood of animals.[Pg 110] Hæmoglobin is a complex compound, consisting of about 96 per cent of albumin (a protein, see Chapter XIII) united with about 4 per cent of hæmatin, a brilliant red pigment which has the formula FeClC32H32O4N4. When treated with acids, the iron (and its accompanying Cl) is removed, and hæmatoporphyrin, C32H36O4N4, is obtained. When either hæmatin, or hæmatoporphyrin is oxidized, hæmatinic acid imide identical with that obtained from ætioporphyrin is obtained. Also, when hæmatoporphyrin is reduced, hæmopyrrole identical with that from ætioporphyrin is obtained. Thus, it would appear that the unit structural groups in hæmatin and in chlorophyll are identical; although chlorophyll may exhibit more variations in isomeric arrangement of these structural units than have been found in hæmatin. Hence, it is apparent that the only essential difference in composition between chlorophyll and hæmatin is that in the former the structural units are linked together by iron, while in the latter, the same units are united through magnesium as the linking element. Further, it is known that while iron is not a constituent element in the chlorophyll molecule, it is, in some unknown way, absolutely essential to the production of chlorophyll in plants; plants furnished with an iron-free nutrient solution rapidly become etiolated and photosynthesis stops.
The following skeleton formulas have been suggested to indicate the way in which these elements are linked between the structural units in their respective compounds.
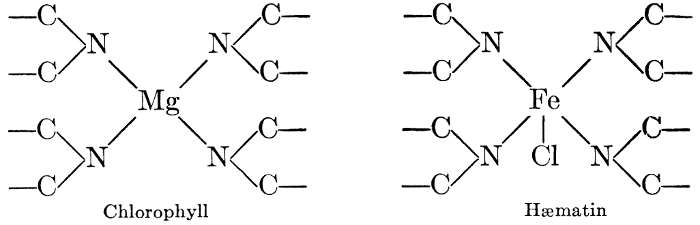
It is understood, of course, that the mineral element does not furnish the definite means of holding the structural units together as otherwise it would not be possible to remove the iron, or magnesium, without breaking down the molecule, as is done in the case of the porphyrins. The actual binding linkage is undoubtedly between carbon atoms, as indicated in Willstätter's formulas for ætiophyllin and ætioporphyrin (see page 109). The attach[Pg 111]ment of the magnesium to each one of the four nitrogen atoms in the skeleton formula assumes the existence of subsidiary valences of 2-4 for magnesium (and of 3-5 for iron), or of possible oscillating valences similar to those supposed to be exhibited by carbon in its closed-ring arrangements.
PROPERTIES OF THE CHLOROPHYLLS
The phytyl esters, or natural chlorophylls, are amorphous solids; while the methylethyl esters (chlorophyllins) and the free acids (phyllins) are crystalline compounds. All of these compounds are easily soluble in ether and alcohol, but insoluble in water. The chlorophylls and chlorophyllins are practically insoluble in petroleum ether and chloroform; but the monobasic acids (pyrrophyllin and phyllophyllin) and the neutral ætiophyllin dissolve easily in chloroform.
Solutions of the chlorophylls are fluorescent, being green by transmitted, and red by reflected light.
Chlorophyll a is a blue-black solid, which gives dark green solutions in all of its solvents. Chlorophyll b is a dark-green solid, which yields brilliant green solutions. Solutions in ether of glaucophyllin and of cyanophyllin are blue; of rhodophyllin, deep violet; of rubiphyllin, light violet; of erythrophyllin, red; and of pyrrophyllin and phyllophyllin, bluish-red. Solutions of the porphyrins are all red, the di-basic ones being usually a bluish-red, and the simpler ones a brilliant red to deep brownish-red in color.
The several chlorophyll derivatives are further distinguished by characteristic differences in their absorption spectra. These differences have been pictured by Willstätter in his book dealing with the results of his investigations concerning the chlorophylls, and reproduced in one or two other texts which treat in detail with the physical-chemical properties of these pigments, but need not be presented in such detail here.
THE CAROTINOIDS
The characteristic brilliant green of healthy plant tissues is due to the fact that there are always associated with the dark bluish-green chlorophylls two (or more) yellow pigments. These[Pg 112] are known as the "carotinoids." This group includes the two brilliant yellow pigments, carotin and xanthophyll, and the reddish brown fucoxanthin and the brilliant red lycopersicin, which are similar in their chemical composition. The first two are found universally distributed in plants, associated with the chlorophylls, and may be regarded as vegetative pigments, although the characteristic ornamental yellow and orange colors of many flowers and fruits, as well as that of the roots of carrots, etc., due to these pigments.
Carotin.—This pigment occurs in various forms in plants, both amorphous and crystalline. It crystallizes out of solution in flat plates, which are orange-red by transmitted light, and greenish-blue by reflected light, and have a melting point of 168°. Carotin is insoluble in water, only very slightly soluble in acetone or cold alcohol, readily soluble in petroleum ether, ether, chloroform, and carbon disulfide. Its solutions are strongly fluorescent.
Its molecular formula is C40H56. It is, therefore, a hydrocarbon of a very high degree of unsaturation. On exposure to dry air, it absorbs 34.3 per cent of its own weight of oxygen, which corresponds to 11-1/2 atoms of oxygen, computed on the basis of the molecular formula C40H56, and would indicate a formula of (C40H56)2O23 for the oxygenated compound; this being three oxygen atoms less than would be required to bring the compound to the theoretical stage of saturation represented by the unimolecular formula CnH2n+2. In moist air, two more oxygen atoms are absorbed, probably forming two OH groups in the molecule. Moreover, carotin absorbs iodine. When the calculated amount of iodine is used, a definite compound having the formula C40H56I2 is produced; but in the presence of an excess of iodine another compound having the apparent formula C40H56I3 (or 2C40H56I2+I2) is obtained. (Note that 2 atoms of iodine plus 12 atoms of oxygen, or 3 of iodine plus 11-1/2 of oxygen, produce the degree of saturation required by the formula CnH2n+2.) It is evident from these experimental data, that a part of the unsaturated linkage in the carotin molecule is of a type which can easily be saturated by direct addition of oxygen, while the remainder may be saturated by iodine.
The reaction of carotin toward bromine is peculiar. With this element, it forms a compound having the formula [Pg 113]C40H36Br22, indicating the direct addition of two atoms of bromine and the substitution of twenty atoms of this element for the same number of hydrogen atoms.
The oxygenated carotins are colorless substances, while the iodide crystallizes in beautiful dark-violet prisms, having a coppery red fluorescence.
Xanthophyll is closely related to carotin. It has the molecular formula C40H56O2. It absorbs 36.55 per cent of oxygen (corresponding to 13 atoms, which would indicate the formation of two OH groups in addition to the saturation required by the CnH2n+2 formula); and an iodine addition product having the formula C40H56O2I2, which crystallizes in dark-violet needles.
Xanthophyll differs markedly from carotin in its solubilities, being insoluble in petroleum ether and only sparingly soluble in carbon disulfide. It may be fairly easily reduced to carotin. This transformation is reversible, and suggests a similarity to the change from hæmoglobin to oxyhæmoglobin, and the reverse, in the blood of animals, as a part of their respiration process.
Separation of the Chlorophylls, Carotin, and Xanthophyll.—These pigments, which exist together in most plant tissues, may easily be separated from each other by taking advantage of the differences in their solubilities, according to the following procedure. Grind up a small quantity of the fresh tissue (leaves of the stinging nettle furnish a conveniently large supply of each of these pigments) with fine sand in a mortar. Cover with acetone, let stand a few moments and then filter on a Büchner funnel. Pour the filtrate into a separatory funnel, add an equal volume of ether and two volumes of water. Shake up once and then allow the ether layer to separate; the pigments will be in this layer. Drain off the water-acetone layer. Now to the etherial solution, add about half its volume of a concentrated solution of potassium hydroxide in methyl alcohol. Shake well and allow to stand until the mixture becomes permanently green. Now add an equal volume of water and a little more ether, until the mixture separates sharply into two layers. The chlorophylls will now be in the lower dilute alcohol layer, and the carotinoids in the upper ether, and may be separated by draining of each layer separately. To separate the carotin from xanthophyll place the ether solution in a small open dish and evaporate to a small volume. Now add about ten volumes of petroleum spirit and an equal volume of methyl alcohol, stir up well, transfer to a separatory funnel and[Pg 114] allow the two layers to separate. The carotin will now be in the upper layer of petroleum ether, and the xanthophyll in the lower alcohol layer; these layers may be drained off separately and the solvents evaporated in order to recover the pigments in dry form.
Lycopersicin (or lycopin) is a hydrocarbon pigment having the same formula as carotin. It is, however, brilliantly red in color, and crystallizes in a different form and has a different adsorption spectrum from carotin. It is the characteristic pigment of red tomatoes, and is found also in red peppers. Yellow tomatoes have only carotin as their skin-pigment, while lycopersicin is usually present in the flesh of the ripe fruits of all varieties and in the skin of red ones. It has been shown, however, that if varieties of tomatoes which are normally red when ripe, are ripened at high temperatures, 90° F. or above, their skins will be yellow instead of red when fully ripe. Hence, the occurrence of carotin, or of lycopersicin, as the skin pigment is determined in part by the varietal character (being different in different varieties when ripened at normal temperatures) and in part by the temperature at which the fruit ripens. The two pigments are, of course, isomers; but the difference in their structural arrangement is not known.
Fucoxanthin, C40H54O6, is a brownish-red pigment, found in fresh brown algæ, and in some brown sea-weeds. Its formula indicates that it is an oxidized carotin. With iodine, it forms a compound having the formula C40H54O6I4. It is unlike carotin and xanthophyll in that it has basic properties, forming salts with acids, which are blue in color.
PHYCOERYTHRIN AND PHYCOPHÆIN
These are the principal pigments of red and brown seaweeds, respectively. Their most characteristic difference from the pigments of non-aquatic plants is that they are easily soluble in water, and insoluble in most organic solvents, such as alcohol, ether, etc. At first thought, this would appear to be impossible, since the plants grow in water and it would seem that their water-soluble pigments would be continuously dissolved out of the tissues. The reason why this does not occur lies in the fact that these pigments exist in the cells of the seaweeds in colloidal[Pg 115] form (see Chapter XV), and, hence, cannot diffuse out through the cell-wails. The only way in which they can be extracted from the tissues is by rupturing the cells, by grinding with sharp sand, etc., after which the pigments can readily be dissolved out by water.
Phycoerythrin is the red pigment. It is a colloidal, nitrogenous substance, allied to the proteins (see Chapter XIII) but not a true protein compound. Hydrolysis by acids indicates that it contains leucine and tyrosine, two amino-acids which are constituents of proteins, along with other bodies of unknown composition.
The colloidal solution of phycoerythrin in water has a brilliant rose-red color, with an orange fluorescence. It readily sets to a gel (see Chapter XV), so that the solution is almost impossible to filter. On this account, purified solutions of this pigment are very difficult to secure, and no satisfactory analysis to indicate its composition has yet been obtained.
Actinically, it is a complementary pigment to chlorophyll, that is, it absorbs the blue and green rays and permits the passage of light which is of the wave length that is absorbed by chlorophyll.
Phycophæin.—Still less is known of the composition of this pigment than of that of phycoerythrin. It is the characteristic pigment of brown seaweeds. It is supposed to exist in the cells of algæ, chiefly as a colorless chromogen, which becomes first yellow and then brown on exposure to air. Associated with it are other pigments, which have been variously reported as carotin, phycoxanthin, etc.
THE ANTHOCYANS
These are a group of pigments of red, blue, or violet color, which occur in the flowers, fruits, or leaves of many species of plants. They are essentially ornamental pigments, and constitute a large proportion of the brilliant colors of flowers, etc. They occur not only dissolved in the cell-sap, but also as deposits of definite crystals or amorphous compounds in the cell protoplasm.
They are all glucosides. When the anthocyans are hydrolyzed, the sugar molecules are split off and the characteristic hydroxy-derivatives of the three-ring anthocyan nucleus (figured on page 83),[Pg 116] known as "anthocyanidins," remain. These anthocyanidins are themselves pigments. They have been shown to be all derivatives of the anthocyan nucleus. The oxygen atom in this nucleus is very strongly basic and exhibits its quadrivalent property by forming stable salts by direct addition of acid radicles. The variation of color of the anthocyanins has been explained by Willstätter, as follows; the red is the acid salt, the blue is a neutral metallic salt, and the violet is the anhydride of the anthocyanidin in question, thus
All of the natural anthocyanin pigments appear to contain a chlorine atom attached directly to the ring oxygen, as shown in the above partial formulas. In addition, they have four, five, or six hydroxyl (OH), or methoxy (OCH3), groups attached at various points around the three rings. The following formula for œnidin, one of the most complex of these anthocyanidins, will illustrate their structural arrangement.
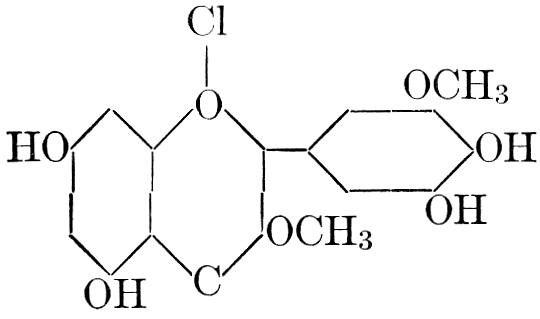
Delphinidin is the corresponding compound without the two CH3 groups; while cyanidin contains only five OH groups; and pelargonidin, only four OH groups.[Pg 117]
The anthocyanin pigments are soluble in water, alcohol, and ether, the solutions being red or blue in color according to the acidity or alkalinity of the medium. Their presence in many species of plants is hereditable, as these plants come true to color from seed, as in the case of red beets, red cabbage, several species of blue berries, etc. In other cases, the anthocyanin development depends largely upon the conditions of growth, particularly those which prevail during the later stages of development: as in the case of apples, where the amount of red color in the skin depends to a large extent upon the conditions under which the fruit ripens.
Anthocyanin pigments often make their appearance late in the season; in fruits, etc., as the result of the normal ripening process but in leaves as the result of shorter daylight illumination accentuated also by sharp frosts.
THE ANTHOXANTHINS
The yellow plant pigments, other than the carotinoids, are almost without exception glucosides having a xanthone or flavone nucleus. These typical nuclei are illustrated on page 83. In these nuclei, as in the anthocyan one, the oxygen atom is strongly basic and combines with mineral acids to form salts (the oxygen becoming quadrivalent) and the color of the pigment depending upon the nature of the combination formed in this way.
The anthoxanthin pigments are yellow, crystalline solids, which are only slightly soluble in water. They dissolve readily in dilute acids and alkalies, giving yellow or red solutions which are of the same color in either acid or alkaline media. They are extensively used as yellow dyes.
Many of the common members of this group have been mentioned in the chapter dealing with the glucosides. The characteristic pigment nucleus of several of these is as follows:
Chrysin, found in various species of poplar and mallows,
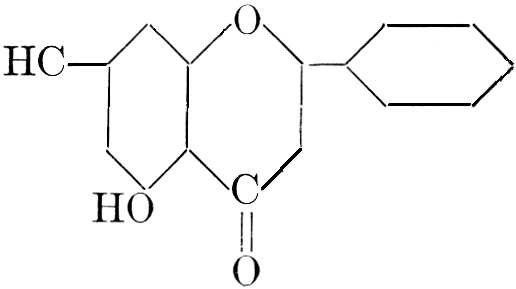
Apigenin, found in parsley and celery, as the glucoside apiin,
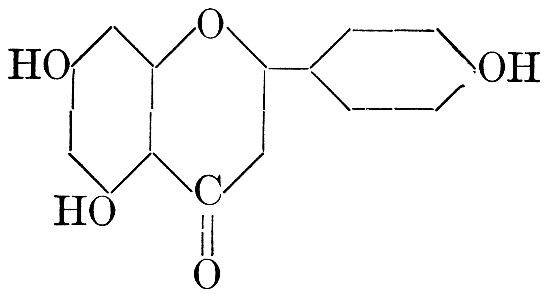
Campferol, found in Java indigo, as the glucoside campferitrin,
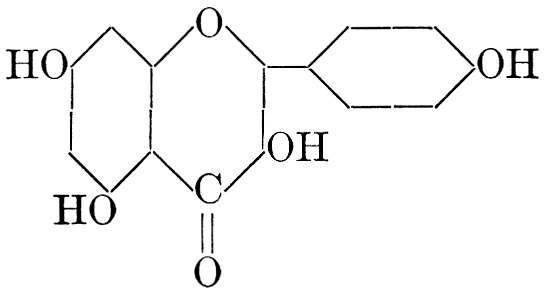
Fisetin, found in quebracho wood and fiset wood,
Quercitrin, found in oak bark, horse-chestnut flowers, and in the skin of onions,
Morin, found in yellow wood (Morus tinctoria).
Gentisin, found in yellow gentian (Gentiana lutea),
As a rule, the most brilliant of these yellow pigments are found in the largest quantities in the bark and wood of various species of tropical plants; although they are also present, in smaller amounts, in the blossoms of species growing in temperate zones.
The anthoxanthins are easily converted into anthocyanins, and vice versa, by the action of oxidizing and reducing enzymes which are commonly present in the tissues of the plants which develop the pigments.
THE PRODUCTION OF ORNAMENTAL PIGMENTS IN FLOWERS, ETC.
The breeding of flowering plants having blossoms of almost any desired color has become a commercial enterprise of large importance. The results which have been obtained, in many cases, have been made the object of scientific study of the genetics of color inheritance. These studies have developed certain interesting facts with reference to the chemistry of the development of these ornamental pigments, which may be briefly mentioned here.
In many of the plants which have been studied, the color of the flowers depends upon several different factors, as follows:
C, a chromogen (or color-producing substance) which is generally a flavone or xanthone glucoside, and which may be either yellow or colorless.[Pg 120]
E, an enzyme which acts upon C, to produce a red pigment.
e, another enzyme which acts upon the red pigment, changing it to some other anthocyanin color.
A, an antioxidase, or antienzyme, which prevents the action of E.
R, an enzyme which changes reds to yellows.
Thus, if a plant whose flower contains only the factor C be crossed with one which contains the factor E, a red blossom will result, or if it contains the factor e more intense pigments are developed. But if either A or R are present, no change in the color of the original parents will result from the crossing.
THE PHYSIOLOGICAL USES OF PIGMENTS
The vegetative pigments undoubtedly serve as agencies for regulating the rate of metabolic processes. At the same time, it is extremely difficult to determine whether the presence of a pigment in any given case is the cause or the effect of the changes in the plant's activities which result from changes in its external environment.
The chlorophylls are, of course, the regulator of photosynthesis, absorbing solar energy with which the photosynthetic process may be brought about. The simultaneous presence of carotinoids in varying amounts undoubtedly serves to modify the amount and character of the radiant energy absorbed, as these pigments absorb a different part of the spectrum of light and hence undoubtedly produce a different chemical activity or "actinic effect" of the absorbed energy. The variations in depth of color of foliage during different growing conditions, from a pale yellow when conditions are unfavorable and growth is slow to the rich dark green of more favorable conditions, is a familiar phenomenon. Whether this change in pigmentation is the result of an adjustment of the plant protoplasm, so that it can absorb a more highly actinic portion of the light, or is a direct effect of the lack of conditions favorable to chlorophyll-production and active photosynthesis, has not yet been determined.
But there must be some influence other than response to environmental conditions which controls the vegetative color in plants, since shrubs, or trees, which have green, yellow, red, and purple leaves, respectively, will grow normally, side by side, under[Pg 121] identical external conditions of sunlight, moisture supply, etc. The hereditary influence must completely overshadow the apparent normal self-adjustment of pigment to energy-absorbing needs, in all such cases.
Again, it appears that there is some definite connection between pigment content and respiration. It is known, of course, that the gaseous exchanges involved in animal respiration are accomplished through the reversible change of hæmoglobin to oxyhæmoglobin, these being the characteristic blood pigments. The easy change of carotin, C40H56, to xanthophyll, C40H56O2, and vice versa, and the reversible changes of the yellow anthoxanthins to the red anthocyanins, under the influence of the oxidizing and reducing enzymes which are universally present in plants, would indicate the possibility of the service of these pigments as carriers of oxygen for respiratory activities in plants in a way similar to that in which the blood pigments serve this purpose in the animal body. The fact, which has been observed in connection with the experimental studies of the development of the lycopersicin, that tomatoes which normally would become red remain yellow in the absence of oxygen, indicates that this pigmentation, at least, is definitely connected with oxygen supply; and the further fact that the development of lycopersicin in red tomatoes, red peppers, etc., is dependent upon the temperature at which the fruit ripens, may indicate a definite connection of this pigment with the need for more oxygen (or for more heat, as suggested in the following paragraph) at these lower temperatures.
Again, many investigators have concluded that at least one function of the anthocyanin pigments is to absorb heat rays and so to increase transpiration and other chemical changes. In support of this view, there may be cited the general presence of such pigments in arctic plants, their appearance in the leaves of many deciduous trees after a frost in the fall, etc. Indeed, there is much to support the view that the autumnal changes in foliage pigments have the physiological function of absorbing heat in order to hasten the metabolic processes of ripening and preparation for winter defoliation. The rapid and brilliant changes in foliage coloring after a sharp frost which kills the tissues and makes rapid translocation of the food material of the leaves to the storage organs immediately necessary, have been explained as the[Pg 122] response of the pigmentation of the leaves to the need for increased heat-absorption. On the other hand, the red pigments of the beet-root, etc., which seem to be identical in composition with the other anthocyanin pigments, can have no such function as those which have just been described. Furthermore, the fact that the pigment often varies in color from red to yellow or brown, depending upon the temperature under which the tissue is ripening, makes it an open question whether the pigment is the regulating agency or whether its nature is the result of the environmental conditions. Or, in other words, it is a question whether these changes in color are a mechanism by which the plant cell adjusts its absorptive powers, or whether they are only the inevitable result of the changes in temperature upon a pigment material which is present in the cell for an entirely different use.
A very interesting side-light upon the color changes which many species of plants undergo when the external temperature falls has been shown by the investigations of the relation of the sugar content of the plant tissues to their pigmentation. It is a well-known fact that not only do many species of deciduous plants show the characteristic reddening of their leaves after frost in the autumn but also many evergreens (Ligustrum, Hedera, Mahonia, etc.) exhibit a marked reddening, or purpling, of their foliage during the winter months, with a return to the normal green color in the spring. Earlier investigations, which have been confirmed by several repetitions, showed that the red or purple leaves always contain higher percentages of sugar than do green ones of similar types. More recent studies have shown that artificial feeding of some species of plants with abnormally large portions of soluble sugars produces a reddening of the foliage tissues which is apparently identical with that which these tissues undergo as the result of low temperatures. Thus, the connection between the natural winter reddening of foliage and the development of sugar in the tissues during periods of low temperatures (see page 64) seems to be clearly demonstrated. It appears that at least a part of the seasonal changes in color of plants is either the cause of, or the effect of, variations in sugar content of the tissues of the plants, accompanying the changes in external temperatures.
Oftentimes, the anthocyanin pigments seem to be associated with sugar production, as contrasted with the chlorophylls, which[Pg 123] seem to be more favorable to the production of starch. But in this case also, it is impossible to say whether the pigment is the direct causative agent in the type of carbohydrate production or whether it is the effect of the same external factors which determine, or modify, the character of the carbohydrate condensation.
BIOLOGICAL SIGNIFICANCE OF ORNAMENTAL PIGMENTS
The ornamental pigments undoubtedly have definite biological significance. When present in the storage roots, such as beet-roots, carrots, etc., or in the above-ground parts of plants, they may have served to protect these organs against herbivorous animals which were accustomed to consume green foods.
In flowers, the brilliant ornamental pigments undoubtedly serve to attract the insects which visit these blossoms in search of nectar, and in so doing promote cross-fertilization. Recent experiments have demonstrated that colors are much more efficient than odors in attracting insects.
Taken altogether, it is apparent that the pigments may have a variety of important rôles in plants. At the same time, some of them may be waste products, with no definite use in the plant economy.
References.
Abderhalden, E.—"Biochemisches Handlexikon, Band 6, Farbstoffe der Pflanzen- und der Tierwelt," 390 pages, Berlin, 1911.
Perkin, A. G. and Everest, A. E.—"The Natural Organic Colouring Matters," 655 pages, London, 1918.
Wakemen, Nellie A.—"Pigments of Flowering Plants," in Transactions of the Wisconsin Academy of Sciences, Arts, and Letters, Vol. XIX, Part II, pages 767-906, Madison, Wisc., 1919.
Watson, E. R.—"Colour in Relation to Chemical Constitution," 197 pages, 65 figs., 4 plates, London, 1918.
Wheldale, M.—"The Anthocyan Pigments of Plants," 304 pages, Cambridge, 1916.
Willstätter, R. and Stoll, A.—"Untersuchung über Chlorophyllen, Methoden und Ergebnisse," 432 pages, 16 figs., Berlin, 1913.[Pg 124]
CHAPTER IX
ORGANIC ACIDS, ACID SALTS, AND ESTERS
Organic acids, either in free form, or partially neutralized with calcium, potassium, or sodium, forming acid salts, or combined with various alcohols in the form of esters, are widely distributed in plants. They occur in largest proportions in the fleshy tissues of fruits and vegetables, where they are largely responsible for the flavors which make these products attractive as food for men and animals. But organic acids and their salts are also found in the sap of all plants, and undoubtedly play an important and definite part in the vital processes of metabolism and growth.
CHEMICAL CONSTITUTION
All organic acids contain one (or more) of the characteristic acid group, —COOH, or 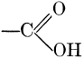 , known as "carboxyl." This group is monovalent, and in the simplest organic acid, formic acid (H2CO2), it is attached to a single hydrogen atom, thus, H·COOH. In all other monobasic acids, it is attached to some other monovalent group, usually an alkyl radical, i.e., a radical derived from an alcohol and containing only carbon and hydrogen (as methyl, CH3, ethyl, C2H5, butyl, C4H9, acryl, C2H3, etc.). Hence, the general formula for all monobasic organic acids is R·COOH, the R representing any monovalent radical. In the simplest dibasic acid, oxalic (H2C2O4), two carboxyl groups are united to each other, thus, HOOC·COOH; but in the higher members of the series, the two characteristic acid groups are united through one or more —CH2— groups, or their oxy-derivatives (as HOOC·CH2·COOH, malonic acid; HOOC·CH2·CH2·CH2·COOH, glutaric acid; HOOC·CHOH·CH2·COOH, malic acid, etc.). Polybasic acids, containing three or more carboxyl groups,[Pg 125] linked together through one or more alkyl carbon atoms, are also possible, and a few typical ones (as
, known as "carboxyl." This group is monovalent, and in the simplest organic acid, formic acid (H2CO2), it is attached to a single hydrogen atom, thus, H·COOH. In all other monobasic acids, it is attached to some other monovalent group, usually an alkyl radical, i.e., a radical derived from an alcohol and containing only carbon and hydrogen (as methyl, CH3, ethyl, C2H5, butyl, C4H9, acryl, C2H3, etc.). Hence, the general formula for all monobasic organic acids is R·COOH, the R representing any monovalent radical. In the simplest dibasic acid, oxalic (H2C2O4), two carboxyl groups are united to each other, thus, HOOC·COOH; but in the higher members of the series, the two characteristic acid groups are united through one or more —CH2— groups, or their oxy-derivatives (as HOOC·CH2·COOH, malonic acid; HOOC·CH2·CH2·CH2·COOH, glutaric acid; HOOC·CHOH·CH2·COOH, malic acid, etc.). Polybasic acids, containing three or more carboxyl groups,[Pg 125] linked together through one or more alkyl carbon atoms, are also possible, and a few typical ones (as
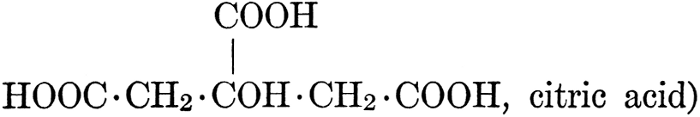
are found in fruits and other plant tissues.
The H atom of the COOH group may be replaced by metals, in exactly the same way as it is replaceable in inorganic acids, producing either neutral or acid salts, depending upon whether all or only a part of the acid H atoms are replaced by the basic element.
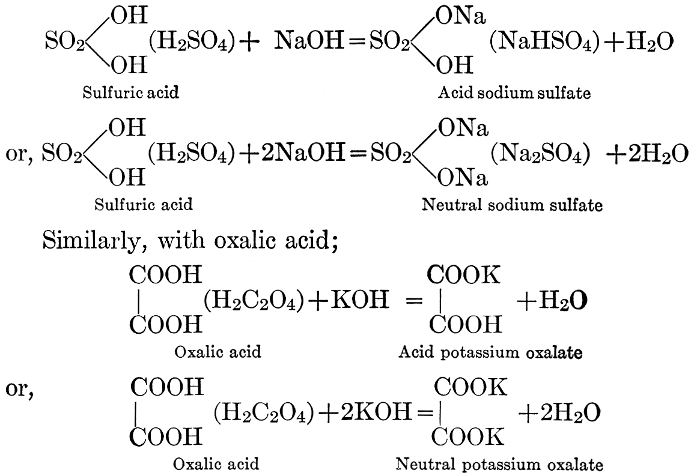
Similarly, the acid H atom of either an organic or an inorganic acid may be replaced by the alkyl group of an alcohol, producing "ethereal salts," or "esters."
Thus, with nitric acid;
| NO2OH (HNO3) | + | C2H5OH | = | NO2OC2H5 (C2H5NO3) | + | H2O |
| Nitric acid | Ethyl alcohol | Ethyl nitrate |
And, with acetic acid;
| CH3·COOH (H4C2O2) | + | C2H5OH | = | CH3·COOC2H5 | + | H2O |
| Acetic acid | Ethyl acetate |
With dibasic or polybasic acids, either one or more of the carboxyl H atoms may be replaced with an alcohol radical, so that[Pg 126] both acid and neutral esters of all such acids are possible. Examples of all of these different types of derivatives of organic acids are frequently found in plant tissues.
The occurrence, properties, and functions of a particular type of glycerol, and other esters of organic acids, which are known as fats and waxes, are not taken into consideration in the following discussions, but reserved for a subsequent chapter dealing specially with them.
SOME COMMON ORGANIC ACIDS
Free organic acids, or their mineral salts or volatile esters, sometimes occur as separate and characteristic individual compounds in particular species of plants, or fruits; but much more commonly, two, three, or even more acids or their derivatives, are associated together.
Formic acid, H·COOH (H2CO2), occurs in free form and in considerable proportions in the leaves of several species of nettle, where it is responsible for the unpleasant effects of the "sting." It may be detected in small amounts in the vegetative parts of many, if not all, plants, especially during periods of rapid growth, and is probably one of the intermediate products in the photosynthesis of carbohydrates (see Chapter III).
Higher members of the formic acid series (as acetic, CH3·COOH; propionic, C2H5·COOH; butyric, C3H7·COOH; etc.) are often found in small quantities in the leaves of many plants and seem to be characteristically present in certain species. They are easily produced from carbohydrates by bacterial action and, hence, are always present in fermenting tissues, such as silage, sauerkraut, etc. Furthermore, the glycerol esters of higher members of this and other monobasic acid series are constituents of all natural fats and oils (see Chapter X).
Oxalic acid, HOOC·COOH (H2C2O4), is found in small amounts in nearly all plants and in relatively large proportions in those of Oxalis, rhubarb, etc. It occurs both as the free acid and as neutral, or acid, oxalates of calcium, potassium, and, perhaps, of magnesium and sodium. Solid crystals of insoluble calcium oxalate are often found in plant cells, and it has been shown that when so deposited the calcium cannot become again available for metabolic uses. It is stated, further, that such[Pg 127] crystals form only when calcium is in excess in the plant sap; hence, the deposition of crystallized calcium oxalate seems to be a device for the avoidance of excessive calcium rather than excessive oxalic acid, in the plant juices.
Succinic acid, HOOC·CH2·CH2·COOH (H6C4O4), occurs in many fruits and vegetables, and is also found in some animal tissues. In fruits, it is usually associated with its derivatives, malic and tartaric acids.
Malic acid, HOOC·CH2·CHOH·COOH (H6C4O5), occurs in apples and in many small fruits, and in many vegetables. Acid calcium malate is now produced commercially as a by-product from the manufacture of syrups from fruit juices, and is used as a substitute for "cream of tartar" in the manufacture of baking powders.
Tartaric acid, HOOC·CHOH·CHOH·COOH (H6C4O6), is found in many fruits, but most characteristically in the grape, where it occurs as the mono-potassium salt. During the fermentation of grape juice into wine, this salt is deposited in considerable quantities in the bottom of the wine-casks. This crude product is collected and sold under the name "argols." From these argols, pure acid potassium tartrate is obtained by decolorization and recrystallization, and constitutes the "cream of tartar" of commerce.
Citric Acid, 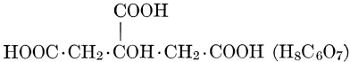 , occurs in large proportions in lemons, and associated with malic acid in strawberries, cherries, currants, etc. It is also found in small quantities in the seeds of the common leguminous vegetables, beans, peas, etc.
, occurs in large proportions in lemons, and associated with malic acid in strawberries, cherries, currants, etc. It is also found in small quantities in the seeds of the common leguminous vegetables, beans, peas, etc.
Tannic acid occurs widely distributed in the plant kingdom as a constituent of the special type of glucosides known as tannins, whose properties and functions have already been discussed (see Chapter VII).
PHYSIOLOGICAL USES OF ORGANIC ACIDS
No conclusive evidence concerning the rôle of organic acids in plant, or animal, growth, has yet been produced. There can be no doubt that the hypothetical carbonic acid and its acid and nor[Pg 128]mal salts have a significant effect in regulating the acidity or alkalinity of plant juices, or body fluids, and so determining the nature of the enzymic activities and colloidal conditions of the biological systems (see Chapters XIV and XV). It is probable that other organic acids, such as formic, acetic, oxalic, and succinic acids, in plants and sarco-lactic acid, in animal tissues, perform similar regulatory rôles; but there seems as yet to be no indication as to why different acids should be used for this purpose by different species, or organisms; or as to the methods by which they perform their specific functions, whatever these may be.
In plants, the organic acids are usually in solution in the sap. When the plant ripens, they generally disappear, either being neutralized by calcium, or other bases, and deposited as crystals in the leaves or stems, or else used up in the synthesis of other organic compounds. Small proportions of these acids are usually present in mature seeds, and the percentage increases materially during germination, indicating that they play an important rôle in insuring the proper conditions for the conversion of the reserve food of the seed into soluble materials available for the nutrition of the young growing plant.
BIOLOGICAL SIGNIFICANCE OF FRUIT ACIDS, ETC.
The occurrence of organic acids, or their derivatives, which have pronounced odors or flavors, in the flesh surrounding the seeds of fruits, in the endosperm of vegetable seeds, or in the tubers, etc., of perennial plants, thus making them attractive as food for animals and men, undoubtedly serves to insure a wider distribution of the reproductive organs of these plants; a fact which has unquestionably had a marked influence upon the survival of species in the competitive struggle for existence during past eras and in the development and cultivation of different species by man. Indirect evidence that the proportion of these attractive compounds present in certain species may have been considerably increased by the processes of "natural selection" in the past is furnished by the many successful attempts to increase the percentage of such desirable constituents in fruits or vegetables by means of artificial selection of parent stocks by skillful plant breeders.[Pg 129]
CHAPTER X
FATS AND OILS, WAXES, AND LIPOIDS
Included in this group are several different kinds of compounds which have similar physical properties, and which, in general, belong to the type of organic compounds known as esters, i.e., alcoholic salts of organic acids. The terms "oil," "fat," and "wax," are generally applied more or less indiscriminately to any substance which has a greasy feeling to the touch and which does not mix with, but floats on, water. There are many oils which are of mineral origin which are entirely different in composition from natural fats. These have no relation to plant life and will not be considered here.
The natural fats, vegetable oils, and plant waxes are all esters. There is no essential difference between a fat and an oil, the latter term being usually applied to a fat which is liquid at ordinary temperatures. The waxes, however, are different in chemical composition from the fats and oils, being esters of monohydric alcohols of high molecular weight, such as cetyl alcohol, C16H33OH, myristic alcohol, C30H61OH, and cholesterol, C27H45OH; whereas the fats and oils are all esters of the trihydric alcohol glycerol, C3H5(OH)3. Lipoids are much more complex esters, having some nitrogenous, or phosphorus-containing, group and sometimes a sugar in combination with the fatty acids and glycerol which make up the characteristic part of their structure.
In general, waxes and lipoids have a harder consistency than fats: but this is not always the case, since "wool-fat" and spermaceti, both of which are true waxes in composition, are so nearly liquid in form as to be commonly called fats; while certain true fats, like "Japan wax," are so hard as to be commonly designated as waxes. It is plain that physical properties alone cannot be relied upon in the classification of these bodies. In fact, there is no single definite property by which members of this group can be accurately identified. There are many other types of substances[Pg 130] belonging to entirely different chemical groups, which have oily, or fat-like, properties.
A. FATS AND OILS
OCCURRENCE
Fats and oils are widely distributed in plants. They occur very commonly in the reproductive organs, both spores and seeds, as reserve food material. In fungi, oils are often found in the spores, but sometimes also in sclerotia, mycelia, or filaments. For example, the sclerotia of ergot have been found to contain as much as 60 per cent of oil. In higher plants, many seeds contain high percentages of oil, so as to make them commercial sources for edible or lubricating oils, such as olive oil, rape-seed oil, cottonseed oil, castor oil, corn oil, sunflower-seed oil, etc., etc. Nuts often contain large proportions of oil, the kernel of the Brazil nut, for example, sometimes contains as high as 70 per cent of oil, while an oil content of 50 per cent, or more, is common in almonds, walnuts, etc.
Oils also occur as reserve food material in other storage organs of plants, such as the tubers of certain flowering plants, and the roots of many species of orchids. Sometimes the appearance of oils in the stems of trees, or the winter leaves of evergreens, seems to be only temporary and to occur only during periods of very low temperatures.
Much less frequently, fats or oils are found in the vegetative organs of plants, as in the leaves of evergreens. Their appearance and functions in these organs seem to be much less certain than in the other cases cited above; although in rare cases a considerable proportion of oily material has been found to exist in definite association with the chloroplasts.
The vegetable fats and oils have many important industrial uses. Some of them, such as olive oil, cottonseed oil, cocoanut oil, etc., are largely used as human food. Others, as castor oil, are used as lubricants. The so-called "drying oils" (see page 132), such as linseed oil, etc., are used in the manufacture of paints and varnishes. Some cheap vegetable oils are used as the basis for the manufacture of soaps, etc. Hence, industrial plants and processes[Pg 131] for the extraction of oils from plant tissues are of very great economic importance.
CHEMICAL CONSTITUTION
The fats (of either plant or animal origin) are glycerides, i.e., glycerol esters of organic acids. As has been pointed out, esters are derived from organic acids and alcohols in exactly the same way that mineral salts are derived from inorganic acids and metallic bases.
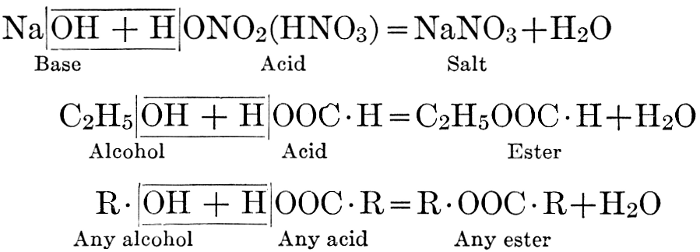
Glycerol is, however, a trihydric alcohol, i.e., it contains three replaceable (OH) groups. Its formula is C3H5(OH)3, or CH2OH·CHOH·CH2OH. Hence, three molecules of a monobasic acid are required to replace all of its (OH) groups.
For example,
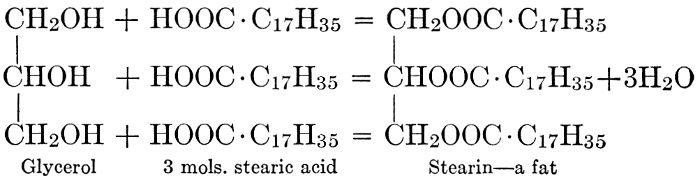
It is theoretically possible, of course, to replace either one, two, or three of the (OH) groups in the glycerol with acid radicals, thus producing either mono-, di-, or triglycerides. If the primary alcohol groups in the glycerine molecule are designated by (1)
and the secondary one by (2), thus, CH2(1)OH·CH(2)OH·CH2(3)OH, it is conceivable that there may be either (1) or (2) monoglycerides, either (1, 1) or (1, 2) diglycerides, or a triglyceride, depending upon which of the (OH) groups are replaced. Compounds of all of these types have been produced by combinations of glycerol with varying proportions of organic acids under carefully controlled conditions; and all of them found to possess fat-like properties.[Pg 132] All natural fats are triglycerides, however. Most natural fats are mixtures of several different triglycerides in each of which the three (OH) groups of the glycerol has been replaced by the same organic acid radical, as in the example of stearin shown above. But recent investigations have shown that some of the common animal fats, and perhaps some plant oils, may be made up of mixed glycerides, i.e., those in which the different (OH) groups have been replaced by different acid groups, as oleo-stearin, oleo-stearo-palmitin, etc.
THE ACIDS WHICH OCCUR IN NATURAL FATS
The acids which, when combined with glycerol, produce fats are of two general types. The first of these are the so-called "fatty acids" having the general formula CnH2n+{1}·COOH. These are the "saturated" acids, i.e., they contain only single-bond linkages in the radical which is united to the ·COOH group; hence, they cannot take up hydrogen, oxygen, etc., by direct addition. The second type are the "unsaturated" acids belonging to several different groups, as discussed below, but all having one or more double-linkages between the carbon atoms of the alkyl radical which they contain. Because of these double linkages, they are all able to take on oxygen, hydrogen, or the halogen elements, by direct addition. When exposed to the air, for example, these "unsaturated" acids, or the oils derived from them, take up oxygen, increasing in weight, and becoming solid or hard and stiff. Hence, natural oils which contain considerable proportions of glycerides of these "unsaturated" acids are known as "drying oils" and are largely used in the manufacture of paints, varnishes, linoleums, etc.; while oils which contain little of these glycerides are known as "non-drying," and are used for food, for lubrication, or for other technical purposes in which it is essential that they remain in unchanged fluid condition when exposed to the air.
The following are some of the more important of the acids which occur as glycerides in natural fats: Saturated Acids:[Pg 133]
- (a) Acetic, or stearic, acid series—general formula, CnH2n+1·COOH.
- (1) Formic acid, H·COOH, occurs free in nettles, ants, etc.
- (2) Acetic acid, CH3·COOH, occurs free in vinegar.
- (3) Butyric acid, C3H7·COOH, in butter fat.
- (4) Capric acid, C9H19·COOH, in butter fat and cocoanut oil.
- (5) Myristic acid, C13H27·COOH, in cocoanut oil and spermaceti.
- (6) Palmitic acid, C15H31·COOH, in palm oil and many fats.
- (7) Stearic acid, C17H35·COOH, in most fats and oils.
Intervening members of this series, such as caprylic acid, C7H15·COOH, and lauric acid, C11H23·COOH, are also found in smaller quantities in cocoanut and palm nut oils, in butter fat, and in spermaceti; while higher members of the series, as arachidic acid, C19H39·COOH, and lignoceric acid, C23H47·COOH, are found in peanut oil; and cerotic acid, C25H51·COOH, and melissic acid, C29H59·COOH, in beeswax and carnauba wax. Unsaturated Acids:
- (b) Oleic acid series—general formula, CnH2n-1·COOH.
- (1) Crotonic acid, C3H5·COOH, occurs in croton oil.
- (2) Oleic acid, C17H33·COOH, occurs in many fats and oils.
- (3) Brassic acid, C21H41·COOH, occurs in rape-seed oil.
- (4) Ricinoleic acid, C17H32OH·COOH, occurs in castor oil.
- (c) Linoleic acid series—general formula, CnH2n-3·COOH.
- (1) Linoleic acid, C17H31·COOH, occurs in linseed and other drying oils.
- (d) Linolenic acid series—general formula, CnH2n-5·COOH.
- (1) Linolenic acid, C17H29·COOH, occurs in many drying oils.
It will be observed that all of these acids contain a multiple of two total carbon atoms. No acid containing an uneven number of carbon atoms has been found in a natural fat. Furthermore, the acids which occur most commonly in natural fats are those which contain eighteen carbon atoms; in fact, more than 80 per cent of the glycerides which compose all animal and vegetable fats are those of the C18 acids. This fact, in addition to the one that the sugars and starches all contain multiples of six carbon[Pg 134] atoms in their molecules, indicates a very great biological significance of the chain of six carbon atoms. This has been alluded to in connection with the discussion of the biological significance of molecular configuration (see page 57) and will be mentioned again in other connections.
THE ALCOHOLS WHICH OCCUR IN NATURAL FATS
Glycerol, as has been pointed out, is by far the most common alcoholic constituent of natural fats and oils. This substance, which is familiar to everyone under its common name "glycerine," is a colorless, viscid liquid having a sweetish taste. It is a very heavy liquid (specific gravity 1.27) which mixes with water in all proportions and when in concentrated form is very hygroscopic.
Glycerine is made from fats and oils by commercial processes which clearly prove that the constitution of fats is as described above. The fat is boiled with a solution of caustic soda and is decomposed, the sodium of the alkali taking the place of the glyceryl (C3H5) group, the latter combining with three (OH) groups from the three molecules of alkali necessary to decompose the fat. A sodium salt of the organic acid, or soap, and glycerol are thus produced, and are separated by saturating the hot solution with common salt, which causes the soap to separate out as a layer on the surface of the liquid, which, on cooling, solidifies into a solid cake, which is then cut and pressed into the familiar bars of commercial soap. From the remaining solution, the glycerine is recovered by evaporation and distillation under reduced pressure. Taking stearin, a common fat, as the example, the reaction which takes place in the above process may be expressed by the following equation:
| C3H5(C17H35·COO)3 | + | 3NaOH | = | 3C17H35COONa | + | C3H5(OH)3 |
| Stearin | Sodium stearate—a soap | Glycerol |
This process, since it yields soap as one of its products, is called "saponification." All fats, when saponified, yield soaps and either glycerol or (more rarely) some of the other alcohols which are described below.
Glycerine is also prepared from fats by hydrolysis with superheated steam. Using olein, a glyceride which is present in olive[Pg 135] oil and many common fats, as the example in this case, the equation for the reaction is:
| C3H5(C17H33·COO)3 | + | 3H2O | = | 3C17H33·COOH | + | C3H5(OH)3 |
| Olein | Steam | Oleic acid | Glycerol |
In this case the free fatty acid, instead of a soap, is the product which is obtained in addition to glycerol.
In the equations presented above, a single glyceride has been used as the example in each case. In the saponification, or hydrolysis, of natural fats and oils which, as has been shown, are mixtures of many glycerides, the resultant soaps, or fatty acids, are mixtures of as many compounds as there were individual glycerides of the original fat, but the glycerol is identical in every case.
When glycerol is heated with dehydrating agents, it is easily converted into acrolein, an unsaturated aldehyde having a peculiar characteristic pungent odor. Hence, the presence of glycerol, or glycerides, in any substance may usually be detected by mixing the material with anhydrous acid potassium sulfate and heating the mixture in a test tube, when the characteristic odor of acrolein will appear.
Glycerol possesses all the characteristic properties of an alcohol, forming alcoholates with alkalies, esters with acids, etc. It is an active reducing agent, being itself easily oxidized to a variety of different products depending upon the strength of the oxidizing agent used and the conditions of the experiment. Microorganisms affect it in a variety of ways, either converting it into simple fatty acids, or condensing it into longer-chain compounds.
Open-chain monohydric alcohols, higher members of the ethyl alcohol series, such as cetyl, C16H33OH, carnaubyl, C24H49OH, ceryl, C26H53OH, and melissyl, C30H61OH, are found in the esters which constitute the major proportion of the common waxes.
Cholesterol and phytosterol are empirical names for certain closed-ring, monohydric alcohols which are found in relatively small amounts in all fats, the former term designating those found in animal fats and the latter those of plant origin. Their composition has not yet been definitely established. They are known to contain two, or three, closed rings, probably of the phenanthrene type; to form dichlor- and dibrom- addition products, showing that they contain one side-chain double linkage; and to yield ketones when oxidized, indicating that they are secondary[Pg 136] alcohols. They form acetyl esters, or acetates, which can be separated from each other and identified by their crystal forms and melting points. Because of this fact and of the further fact that they are present in detectable quantities in practically all fats and oils, they afford a qualitative means of distinguishing between fats of animal and of plant origin. This possibility is the most interesting fact known concerning these complex alcohols; although their presence as esters in all plant and animal fats indicates that they must have some biological function.
Phytosterol is not a single alcohol, but a mixture of at least two, which have been separated and studied as sitosterol, C27H43OH, and stigmasterol, C30H49OH. As has been said, these are found in small proportions in all vegetable fats, being present in largest amounts in oily seeds, especially those of the legumes.
The saponification of esters of cholesterol and phytosterol is a difficult and unsatisfactory process; but since this affords the only known means to distinguish between fats of plants and of animal origin, its technique has been fairly well worked out, and the process used in the study of the changes which take place in plant fats when they are used by animals as food.
HYDROLYSIS AND SYNTHESIS OF FATS
The reaction for the hydrolysis of fats has been discussed in connection with the process for the manufacture of glycerine. This reaction takes place very slowly with cold water alone, can be easily brought about by the action of superheated steam, and much more easily and rapidly in the presence of some catalyst (sulfuric acid is an especially effective catalyst for this purpose).
Fats can be artificially synthetized by heating mixtures of glycerol and fatty acids, under considerable pressure, for some time at temperatures of 200° to 240° C.; or by heating a mixture of the disulfuric ester of glycerol with a fatty acid dissolved in sulfuric acid. Recently, fatty acids have been prepared from carbohydrates, by first breaking the hexoses down into three-carbon compounds, then carefully oxidizing these to pyruvic acid, CH3·CO·COOH, which can then be condensed into acids having longer chains. The violent reagents and long-continued processes which must be employed for the artificial hydrolysis or synthesis of the fats are in sharp contrast with the easy and rapid transition[Pg 137] of carbohydrates to fats, and vice versa, which take place in both plant and animal nutrition.
THE EXTRACTION OF OILS FROM PLANT TISSUES
There are three types of methods which are employed for the extraction of oil from oil-bearing seeds, etc., either as a commercial industry or for the purposes of scientific study. These are (1) by pressure; (2) extraction with volatile solvents; and (3) boiling the crushed seeds or fruits with water.
By the first method, the seeds are first cleaned, then "decorticated" (hulls removed), crushed or ground, then subjected to intense pressure in an hydraulic press. In the commercial process, the ground seeds are first pressed at ordinary temperature, which yields "cold-drawn" oil, then the press cake is heated and pressed again, whereby "hot-drawn" oil is obtained. The crude oil is refined by heating it to coagulate any albumin which it may contain, and is sometimes bleached by different processes before it is marketed. The press cake from many seeds, such as flaxseed (linseed), cottonseed, etc., is ground up and sold for use as stock feed.
In the second method, the finely crushed seeds are treated with solvents such as gasoline or carbon bisulfide, in an apparatus which is so arranged that the fresh material is treated first with solvent which has already passed through various successive lots of material and has become highly charged with the oil, followed by other portions which contain less oil, and finally by fresh solvent, whereby the last traces of oil are removed from the material. The saturated solvent is transferred to suitable boilers and the solvent distilled off and condensed for repeated use, leaving the oil in the boiler in very pure form.
Extraction by boiling with water is sometimes used in the preparation of castor oil and olive oil. In such cases, the crushed seeds are boiled with water and the oil skimmed off as fast as it rises to the surface.
IDENTIFICATION OF FATS AND OILS
Fats and oils are identified by determinations of their physical properties, such as specific gravity, melting point, refractive[Pg 138] index, etc., and by certain special color reactions for particular oils; or by measurements of certain chemical constants, such as the percentage of free fatty acids which they contain, the saponification value (i.e., the number of milligrams of KOH required to completely saponify one gram of the fat), the iodine number (percentage by weight of iodine which is absorbed by the unsaturated fatty acids present in the fat), percentage of water-insoluble fatty acids obtained after saponification and acidifying the resultant soap, etc., etc. Most of these tests must be carried out under carefully controlled conditions in order to insure reliable identifications, and need not be discussed in detail here. Full directions for making such tests, together with tables of standard values for all common fats and oils, may be found in any reference book on oil analysis.
PHYSIOLOGICAL USE OF FATS AND OILS
In animal organisms, fats are the one important form of energy storage. They also form one of the most important supplies of energy reserve material in plants. Carbohydrates commonly serve this purpose in those plants whose storage reservoirs are in the stems, tubers, etc.; but in most small seeds the reserve supply of energy is largely in the form of oil, and even in those seeds which have large endosperm storage of starch, the embryo is always supplied with oil which seems to furnish the energy necessary for the first germinative processes.
Fats are the most concentrated form of potential energy of all the different types of organic compounds which are elaborated by plants. This is because they contain more carbon and hydrogen and less oxygen in the molecule than any other group of substances of vegetable (or animal) origin. It has been pointed out that a quantity of fat capable of yielding 100 large calories of heat will occupy only about 12 cc. of space, whereas from 125 to 225 cc. of space in the same tissue would be required for the amount of starch of glycogen necessary to yield the same amount of heat, or energy, when oxidized.
The fats undoubtedly catabolize first by hydrolysis into glycerol and fatty acids, and then by oxidation possibly first into carbohydrates and then finally into the end-products of oxidation, namely, carbon dioxide and water. The following hypothetical[Pg 139] equation to represent the oxidation of oleic acid into starch, suggested by Detmer, is interesting as a suggestion of how much oxygen is required and how much heat would be liberated by such a transformation:
C18H34O2 + 27O = 2(C6H10O5) + 6CO2 + 7H2O
Complete oxidation of oleic acid to the final end-products, carbon dioxide and water, would require much more oxygen, thus:
C18H34O2 + 51O = 18CO2 + 17H2O.
Hence, Detmer's reaction would yield only approximately one-half the total energy available in the acid; but it does indicate the possibility of redevelopment of fatty acids or fats from the unoxidized carbohydrate material which remains in the equation. Moreover, there is abundant evidence to show that, in both animal and plant tissues, energy changes are brought about chiefly by the transformation of fats into carbohydrates and vice versa.
Many different hypotheses have been put forward concerning the mode of transformation of fats into carbohydrates, and the changes which take place in oily seeds during their germination have been carefully studied by many investigators. The following seem to be fairly well established facts. First, that fats as such may be translocated from cell to cell, since cell-walls and cell protoplasm seem to be permeable to oil if it is a sufficiently fine emulsion; or they may be hydrolyzed into glycerol and fatty acids and translocated from cell to cell in these forms and recombined into fats in the new location. Second, that fats are formed from glucose in some plants, from sucrose and from starch in others, and from mannite and similar compounds in still other species. Third, that in germination the fatty acids are used up in the order of their degree of unsaturation, those which contain the largest number of double-bond linkages being used first, and the saturated acids last of all. Fourth, that the sugar produced by the oxidation of fats is derived either from the glycerol or from the fatty acids of the fat, depending upon the nature of the latter. If the fat is saturated, the glycerine is converted into sugar while the fatty acids are oxidized; but if the fat contains large proportions of unsaturated acids, these contribute to the formation of sugar.[Pg 140]
Recent studies seem to show that in the animal body fats serve an important function in connection with the production of antibodies to disease germs. But there is as yet no evidence to show that fats and oils have any similar function in plant tissues. The fact that they are found almost wholly in the storage organs of plants seems to indicate that their use as food reserve material is their principal, if not their sole, function in the plant economy.
B. THE WAXES
Waxes are most commonly found in or on the skin of leaves or fruits. They are similar to fats in chemical composition, except that, instead of being glycerides, they are esters of monohydric alcohols of high atomic weight. The term wax, when used in the chemical sense, has reference to this particular type of esters rather than to any special physical properties which the compound possesses, and both solid and liquid waxes are known.
Carnauba wax, found on the leaves of the wax-palm (Copernicia cerifera) contains ceryl alcohol (C23H53OH) and myricyl alcohol (C30H61OH) esters of cerotic acid (C25H51·COOH) and carnaubic acid (C23H47·COOH). It is the best known vegetable wax. Poppy wax is composed chiefly of the ceryl ester of palmitic acid (C17H35·COOH).
Since waxes contain no glycerol, they give no odor of acrolein when heated with dehydrating agents, do not become rancid, and are less easily hydrolyzed than the fats. They are soluble in the same solvents as the fats, but generally to a less degree.
The facts that waxes are impervious to water and usually occur on the surfaces of plant tissues have led to the conclusion that their chief function is to provide against the too-rapid loss of water by evaporation from these tissues. This seems to be borne out by the common experience that many fresh fruits and vegetables will keep longer without shriveling if their waxy coating is undisturbed. No other function than that of regulation of water losses has been suggested for the plant waxes.
C. THE LIPOIDS
The lipoids, or "lipins," as some authors prefer to call them, are substances of a fat-like nature which are found in small quantities in nearly all plant and animal tissues and in considerable[Pg 141] proportions in nerve and brain substance, in egg yolk, etc., and in the seeds of plants. When hydrolyzed, they yield fatty acids or derivatives of fatty acids and some other group containing either nitrogen only or both nitrogen and phosphorus. The facts that they are extracted from tissues by the same solvents which extract fats and that they yield fatty acids when hydrolyzed account for the name "lipoid," which comes from the Greek word meaning fat. Some writers, who object to the word "lipoid" as a group name, prefer to call these substances the "fat-like bodies."
The first group of lipoids to be studied were those which occur in the brain; and the name cerebroside was given to those lipoids which, when hydrolyzed, yield fatty acids, a carbohydrate and a nitrogen-containing compound but no phosphoric acid; while those lipoids which contain both nitrogen and phosphorus were called phosphatides. Substances which correspond in composition to both these types are found in plant tissues and the same class names are applied in a general way to lipoids of either plant or animal origin.
Plant lipoids have not been studied to nearly the same extent as have those which occur in the animal body; and certain observers believe that there are significant differences between the lipoids of plants and those of animal origin. However, most investigators use the same methods of study and the same systems of nomenclature for these fat-like substances, regardless of their origin.
LECITHIN
This phosphatide is by far the best-known lipoid. It occurs in the brain, the heart, the liver, and in the yolk of the eggs of many animals; and either lecithin or a substance so nearly like it in character as to be regarded by most investigators as identical with it, is present in small, but constant, quantities in nearly all seeds, especially those of leguminous plants. In many legume seeds, it constitutes from 50 to 60 per cent of the "ether extract," or "crude fat," which can be extracted from the crushed seeds, using ether as the solvent.
Lecithin is a glyceride. Only two of the (OH) groups of the glycerol are replaced by fatty acids, however; the third being replaced by phosphoric acid, H3PO4, or PO(OH)3, which, in turn, has one of its hydrogen atoms replaced by the base choline. Choline is a nitrogenous base, or amine, which may be regarded as[Pg 142] ammonium hydroxide with three of its hydrogen atoms replaced by methyl groups and the fourth by the ethoxyl group, the latter being the ethyl group with an OH in place of one of its hydrogens. Thus,

Without the choline, lecithin would be a di-fatty acid derivative of glycero-phosphoric acid. These relations may be seen in the following formulas:
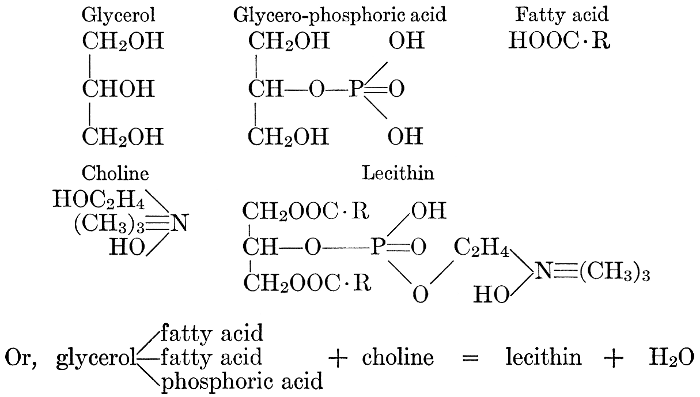
There are many different possible linkages of the constituent groups which make up the lecithin molecule. In the first place, if the (OH) groups of the glycerol molecule be numbered (1) and (2), thus,
the fatty acid radicals may be attached either in one (1) position and one (2) position, or in the two (1) positions; hence, two forms of glycero-phosphoric acid are possible, thus
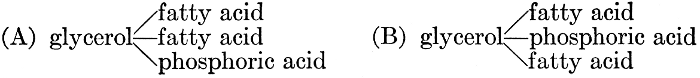
Again, the choline may be attached to the phosphoric acid either through its alcoholic (OH) group or through its basic (N) group, thus
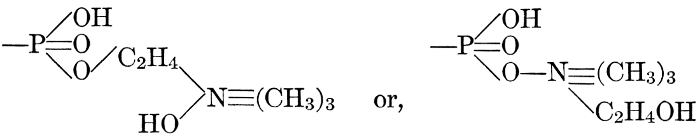
The facts that in the arrangement (B) the central carbon atom of the glycerol would be asymmetric, and that both lecithin and the glycero-phosphoric acid derived from it by hydrolysis are optically active, prove that formula (B) correctly represents the arrangement of that part of the lecithin molecule; and there is ample theoretical and experimental evidence to prove that the choline linkage is through the alcoholic (OH) group. Hence the formula for lecithin indicating the linkage as shown above is the correct one.
The fatty acids in the lecithin molecule may be different in lecithins from different sources, just as they are different in fats from different sources. Both oleic acid and a solid fatty acid have been found in the hydrolysis products of lecithin from leguminous seeds. In certain lupine seeds, the fatty acids present in the lecithin appear to be palmitic and stearic.
OTHER PLANT PHOSPHATIDES
Phosphatides other than lecithin are common in plants. In these, various sugars replace part or all of the glycerol as the alcoholic part of the ester. Percentages of sugar varying from mere traces up to 17 per cent of the weight of material taken, have been found in the products of hydrolysis of phosphatides prepared from vetch seeds, potato tubers, plant pollens, and whole wheat meal.
Furthermore, betaine
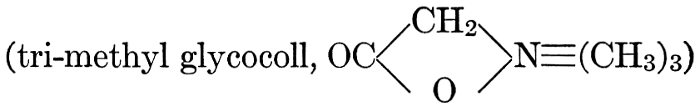
and perhaps other vegetable amines (see Chapter XII) sometimes replace choline as the basic group in the phosphatides.[Pg 144]
PLANT CEREBROSIDES
Bodies similar to the animal cerebrosides seem to occur in many plant tissues, since plant lipoids which yield no phosphorus when hydrolyzed have often been isolated. The sugar which constitutes the alcoholic portion of their structure appears to be galactose in every case which has been reported. Beyond this, little is known of the structure of these plant cerebrosides, as they are very difficult to prepare in pure form and not easily hydrolyzed.
PHYSIOLOGICAL USES OF LIPOIDS
Lipoids are so universally present in plant and animal tissues and so commonly found in those parts of the organism in which vital phenomena are most pronounced (brain, heart, embryo of egg, embryo of seeds, etc.), that it is evident that they must play some important rôle in the activity of living protoplasm. There is, as yet, however, no definite and certain knowledge of what this rôle is. Various theories concerning the matter have been put forward in recent years. For example, Overton, in 1901, presented the idea that every living cell is surrounded by a semi-permeable membrane consisting of lipoid material, which regulates the passage into and out of the cell of substances necessary to its metabolism and growth. Recent investigations by Osterhout and others indicate, however, that Overton's hypothetical lipoid membrane is not essential to a proper explanation of the migration into and out of the cell protoplasm of nutritive materials, etc. Other investigators have cited results which appear to indicate that lipoids play an important, but as yet unknown, part in the process of fat metabolism. Others go even further than this, and argue that since the extraordinary rapidity of the chemical changes which take place in plant protoplasm indicates the necessity of the presence there of exceedingly labile substances, and since both fats and proteins are relatively stable compounds, it is possible that the lipoids, which contain both nitrogenous and fatty acid groups, play an exceedingly important part in the metabolism processes. Bang, in particular, has pointed out (in 1911) that the lipoids are probably the most labile of all the components which constitute the colloidal system known as plant protoplasm. The importance of such considerations will be more apparent after the[Pg 145] relation of colloidal phenomena to the activities of plant cell contents has been more fully discussed (see Chapter XVI).
Experimental studies of the physiological uses of lipoids have thus far been devoted almost exclusively to those of animal tissues. They have been seriously hampered by the difficulty of securing properly purified extracts of lecithin and similar lipoids. The same labile character which apparently makes them so important in the chemical changes in the cell makes them equally unstable compounds to work with in attempting to secure pure preparations for the purposes of experimental study. On this account, there is, as yet, no certain knowledge concerning their actual physiological uses. It is evident, however, that they have some really important rôle to play, which opens up a promising field for further study.
References.
Abderhalden, E.—"Biochemisches Handlexikon, Band 3, Fette, Wachse, Phosphatide, Cerebroside, ..." 340 pages, Berlin, 1911.
Hopkins, E.—"The Oil-Chemist's Handbook," 72 pages, New York, 1902.
Leathes, J. B.—"The Fats," 138 pages, Monographs on Biochemistry, London, 1913.
Lewkowitsch, J.—"Chemical Technology and Analysis of Oils, Fats, and Waxes," Vol. I, 542 pages, 54 figs.; Vol. II, 816 pages, 20 figs.; and Vol. III, 406 pages, 28 figs., London, 1909.
Maclean, H.—"Lecithin and Allied Substances," 206 pages, Monographs on Biochemistry, London, 1913.
Southcombe, J. E.—"Chemistry of the Oil Industries," 204 pages, 13 figs., London, 1918.[Pg 146]
CHAPTER XI
ESSENTIAL OILS AND RESINS
Included in this group are all those substances to which the characteristic odors of plants are due, along with others similar in structure and possessing characteristic resinous properties. They have no such uniformity in composition as is exhibited by the oils which are included among the fats and waxes; but belong to several widely different chemical groups. Furthermore, there is no sharp dividing line between the essential oils and certain esters of organic acids on the one hand and the fats on the other. For example, if an aromatic fluid essence is a light fluid, non-viscid, and easily volatile, it is usually classed with the organic esters; denser liquid substances, of oily or waxy consistency, and with comparatively slight odor and taste are usually fats, while oils of similar physical properties but possessing strong characteristic odors are classed as essential oils, regardless of their chemical composition.
Included in this general class are compounds having a great variety of chemical structures; e.g., hydrocarbons, alcohols, phenols, organic sulfides and sulfocyanides, etc. Many of these compounds are crystalline solids at ordinary temperatures, but melt to oily fluids at higher temperatures. The characteristic property which assigns any given plant extract to this group is that it has a strikingly characteristic odor or taste, often accompanied by some definite physiological effect, or medicinal property.
These compounds may be either secretions or excretions of plants, sometimes normally present in the healthy tissue, and sometimes produced as the result of injury or disease.
The essential oils and the resins often occur associated together in the plant; or, the resins may develop from the oily juice of the plant after exposure to the air.[Pg 147]
THE ESSENTIAL OILS
These may be divided, according to their chemical composition, into two major groups; (1) the hydrocarbon oils, or terpenes, and (2) the oxygenated and sulfuretted oils.
The terpenes are of three different types, namely: (a) the hemiterpenes, C5H8, unsaturated compounds of the valerylene series, of which isoprene (found in crude rubber) is the best-known example; (b) the terpenes proper, C10H16, which constitute the major proportion of the whole group; and (c) the polyterpenes (C5H8)n, of which colophene and caoutchouc are the most common examples.
Eleven different terpenes having the formula C10H16 have been isolated from various plant juices, and their molecular arrangement carefully worked out. The following three examples will serve as typical of the general structural arrangement of these hydrocarbons:
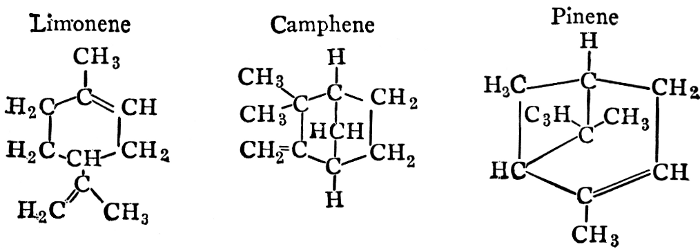
A discussion of the evidence which supports these formulas as properly represented the molecular arrangements of the various isomeric forms would be out of place here, as its only particular interest is in connection with the medicinal effects of the different compounds. It is clear, however, that they are six-membered hydrocarbon rings, with additional hydrocarbon groups attached to one or more of the carbon atoms in the ring.
Different modifications, or varieties, of the terpenes constitute the main proportions of the oils of turpentine, bergamot, lemon, fir needles, eucalyptus, fennel, pennyroyal, etc.
The oxygenated essential oils may be either alcohols, aldehydes, ketones, acids, esters, or phenols, derived from either five-membered or six-membered closed-ring hydrocarbons. They are usually present in the plant oil in mixtures with each other or with a terpene. Since most of them have pronounced physiological[Pg 148] or medicinal properties, their structure has been well worked out, in most cases; but it seems to be hardly worth while to present these matters in detail here, as they are of interest chiefly on account of their medicinal properties rather than their botanical functions.
Borneol, C10H17OH, and menthol, C10H19OH, are typical alcohols. The latter is a crystalline substance, which melts at 42°, which is present in peppermint oil, both as the free alcohol and as an ester of acetic acid.
Amyl acetate, CH3·COOC5H11, and linalyl acetate, CH3·COOC10H17, the latter occurring in the oils of lavender and bergamot, are typical esters classed as essential oils.
As examples of the aldehyde oils, benzoic aldehyde, C6H5CHO, "oil of bitter almonds," and cinnamic aldehyde, C6H5CH=CHCHO, found in the oils of cinnamon and cassia, may be cited.
Camphor, C10H16O, is a ketone, having the following structural formula:
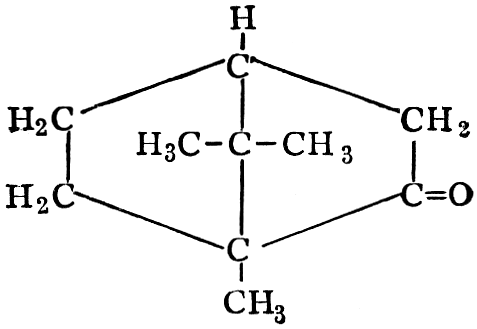
There are a considerable number of essential oils which are phenols. Thymol, C6H3·(CH3)·(C3H7)·OH, in oil of thyme, and carvacrol, its isomer, in oil of hops, are familiar examples.
Coumarin, the anhydride of cinnamic acid,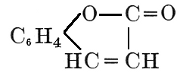 ; is an example of an acid substance which is classed as an essential oil, even though it is a solid at ordinary temperatures. It has an odor and flavor similar to that of vanillin, the essential flavoring material of the vanilla bean, and is often used as a substitute for the latter in the preparation of artificial flavoring extracts.
; is an example of an acid substance which is classed as an essential oil, even though it is a solid at ordinary temperatures. It has an odor and flavor similar to that of vanillin, the essential flavoring material of the vanilla bean, and is often used as a substitute for the latter in the preparation of artificial flavoring extracts.
Of the essential oils containing sulfur, there are two common examples; oil of mustard, allyl isosulfocyanide, C3H5NCS, and oil of garlic, allyl sulfide (C3H5)2S. The latter is present in onions, garlic, water cress, radishes, etc., the difference in flavor of these vegetables being due to the fact that the allyl sulfide is united with other different groups in the glucoside arrangement,[Pg 149] in the different plants. Similarly, mustard oil is not present in mustard seeds as such, but as a glucoside which, when hydrolyzed by the enzyme myrosin which is always present in other cells of the same seeds, yields C3H5NCS, KHSO4, and C6H12O6.
THE RESINS
The resins were formerly supposed to be the mother substances from which the terpenes are derived. It is now known, however, that they are the oxidation products of the terpenes. Their exact structure is still a matter of some uncertainty, as their peculiar "resinous" character makes them very difficult to study by the usual methods of chemical investigations.
Resins are divided into two classes: (a) the balsams, and (b) the solid or hard resins. Canada balsam and crude turpentine are familiar examples of the first class. They consist of resinous substances, dissolved in or mixed with fluid terpenes. Ordinary resin, or colophony, consists chiefly of a monobasic acid having the empirical formula C20H30O2, known as sylvinic acid, whose exact structure is not known. Its sodium salt is used as the basis for cheap soaps.
The hard resins are amorphous substances of vitreous character, which consist of very complex aromatic acids, alcohols, or esters, combined with other complicated structures, known as resenes, whose definite chemical nature is not yet known. Among the hard resins are many substances which are extensively used in the manufacture of varnishes, such as copal, amber, dammar, sandarach, etc.
There are also resinous substances, such as asafœtida, myrrh, gamboge, etc., which are mixtures of gums (see Chapter VI) and true resins. Some of these have considerable commercial value for medicinal or technical uses.
PHYSIOLOGICAL USES AND BIOLOGICAL SIGNIFICANCE OF ESSENTIAL OILS
No theory has yet been advanced concerning the possibility of the use of essential oils and resins by plants in their normal metabolic processes. The very great diversity in their chemical nature makes it impossible that they should all be considered as having[Pg 150] the same physiological function, if indeed any of them actually have any such function.
It is evident that those aromatic compounds which occur as normal secretions of plants and which give to the plants their characteristic odors may act either as an attraction to animals which might utilize the plants as food and so serve to distribute the seed forms, or as a repellent to prevent the too rapid destruction of the leaves, stems, or seeds of certain species of plants whose slow-growing habits require the long-continued growth of these portions of the plant for the perpetuation of the species. The presence of these compounds in larger proportions in those species of conifers, etc., which grow in tropical regions, in competition with other rapid-growing vegetation, suggests the latter possibility. It must be admitted, however, that their presence in such cases may be the result of climatic conditions, as indicated by the fact that most spice plants are tropical in habit, rather than the result of their protective influence in the struggle for survival during past ages.
Many of the oils and resins which are secreted as the result of injury by disease or wounds have marked antiseptic properties and undoubtedly serve to prevent the entrance into the injured tissue of destructive organisms.
But apart from these possible protective influences which may have had an important effect upon the preservation and perpetuation of the species of plants which secrete them, there is no known biological necessity for the presence of these aromatic substances in plants.
References.
Abderhalden, E.—"Biochemisches Handlexikon, Band 7, Gerbstoffe, Flechtenstoffe, Saponine, Bitterstoffe, Terpene, Aetherische Oele, Harze, Kautschuk," 822 pages, Berlin, 1912.
Allen's Commercial Organic Analysis, Vol. IV, "Resins, Rubber, Guttapercha, and Essential Oils," 461 pages, 7 figs., Philadelphia, 1911 (4th ed.).
Heusler, F., trans. by Pond, F. J.—"The Chemistry of the Terpenes," 457 pages, Philadelphia, 1902.
Parry, E. J.—"The Chemistry of Essential Oils and Perfumes," 401 pages, 20 figs., London, 1899.[Pg 151]
CHAPTER XII
THE VEGETABLE BASES
We come, now, to the consideration of the characteristically nitrogenous compounds of plants. None of the groups of compounds which have been considered thus far have, as a group, contained the element nitrogen. This element is present in the chlorophylls and in certain other pigments, but not as the characteristic constituent of the molecular structure of the group of compounds, nor do these compounds serve as the source of supply of nitrogen for the plant's needs.
The characteristic nitrogen-containing compounds may all be regarded as derived from ammonia, or ammonium hydroxide, by the replacement of one or more hydrogen atoms with organic radicals of varying type and complexity. If the group, or groups, which be considered as having replaced a hydrogen atom in ammonia, in such compounds, is an alkyl group, the compound is strongly basic in character and is known as an amine; whereas if the replacing group is an acid radical, the resulting compound may be neutral (known as acid amides), or weakly acid (known as amino-acids) in type. Compounds of the first type constitute the vegetable bases; while those of the second type are the proteins.
The vegetable bases may be divided into three groups. These are (a) the plant amines, which are simple open-chain amines; (b) the alkaloids, which are comparatively simple closed-ring amines, containing only one nitrogen atom in any single ring; and (c) the purine bases, which are complex compounds containing a nucleus with four carbon atoms and four nitrogen atoms arranged alternately to form a double-ring group.
THE PLANT AMINES
The simple amines bear the relation to ammonia, or ammonium hydroxide, represented by the following formulas, in which the R indicates any simple alkyl radical:[Pg 152]
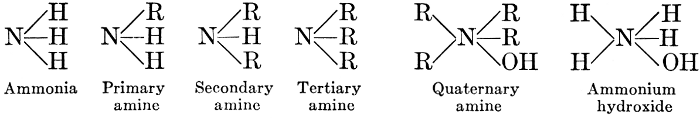
The simple amines which occur in animal tissues are known as "ptomaines" and "leucomaines." The ptomaines are all decomposition products resulting from the putrefactive decay of proteins caused by moulds or bacteria. Some of these are highly toxic, producing the so-called "ptomaine-poisoning"; while others are wholly innocuous. They are all simple amines. Putrescine, di-amino butane, NH2·CH2·CH2·CH2·CH2·NH2, and cadaverine, di-amino pentane, HN2·(CH2)5·NH2, are common non-toxic ptomaines, resulting from the decay of meat. Neurine, trimethyl-ethylene ammonium hydroxide, (CH3)3(C2H3)·NOH, is a violently poisonous ptomaine produced in the decay of fish. Amines of similar structure to these are occasionally found in living animal tissues. Such compounds are known as leucomaines, to distinguish them from the ptomaines, which are found only in dead material.
Corresponding in structure and properties to these amines of animal origin, there is a series of basic substances, found in many plants, known as the plant amines. The following are common examples:
Trimethyl amine, (CH3)3N, is a very volatile compound, found in the flowers of several species of the Rose family, the leaves of certain weeds, etc. When crushed, these tissues give off a very fetid odor, which is due to this amine.
Choline, muscarine, and betaine are plant amines which are closely related to each other and to neurine (the toxic ptomaine) in composition and structure, as shown in the following formulas:
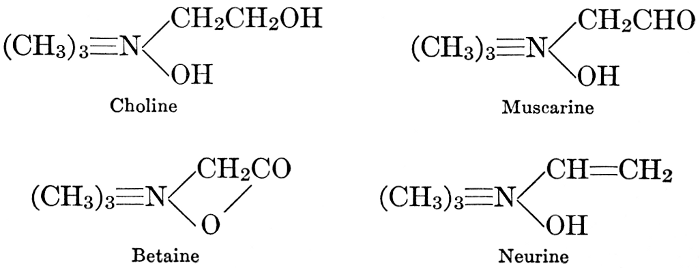
Choline and betaine are non-toxic; while muscarine and neurine are violent poisons.
Choline and muscarine occur in certain toadstools. Betaine and choline often occur together in the germs of many plants. Betaine is found in the beet root and the tubers of Jerusalem artichoke. Choline occurs alone in the seeds and fruits of many plants, sometimes as the free amine, but more often as a constituent of lecithin (see page 141).
Phenyl derivatives of simple amines are sometimes found in plants. Hydroxyphenylethyl amine,
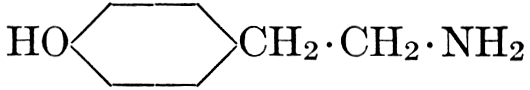
found in ergot, and hordein,
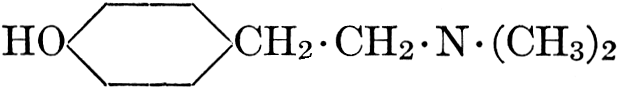
found in barley, are examples. The former has marked medicinal properties.
There is no known physiological use for these simple amines in plants. By some investigators, they are regarded as intermediate products in the synthesis or decomposition of proteins; but it would seem that if this were a normal procedure, these amines would occur in varying proportions in all plants, under different conditions of metabolism, instead of in practically constant proportions in only a few species, as they do.
ALKALOIDS
These are a group of strong vegetable bases whose nitrogen atom is a part of a closed-ring arrangement.
As a rule, alkaloids are colorless, crystalline solids, although a few are liquids at ordinary temperatures. They are generally insoluble in water, but easily soluble in organic solvents. Being strong bases, they readily form salts with acids, and these salts are usually readily soluble in water.
Alkaloids are usually odorless; although nicotine, coniine, and a few others, have strong, characteristic odors. Most of them have a bitter taste, and many of them have marked physiological effects upon animal organisms, so that they are extensively used as narcotics, stimulants, or for other medicinal purposes.
Most of the alkaloids contain asymmetric carbon atoms and are, therefore, optically active, usually levorotatory, although a few are dextrorotatory.[Pg 154]
The alkaloids are precipitated out of their solutions by various solutions of chemical compounds, known as the "alkaloidal reagents": iodine dissolved in potassium iodide solution gives a chocolate-brown precipitate; tannic acid, phosphotungstic acid, phosphomolybdic acid, and mercuric iodide solutions give colorless, amorphous precipitates; while gold chloride and platinic chloride solutions give crystalline precipitates, many of which have sharp melting points and can be used for the identification of individual alkaloids. There are a great many specific color reactions for individual alkaloids, which are important to toxicologists and pharmacists, but which it would not be desirable to consider in detail here.
The alkaloids are conveniently divided into groups, according to the characteristic closed-ring arrangements which they contain. The several closed-ring arrangements which are found in common alkaloids, and upon which their grouping is based, may be illustrated by the following formulas:
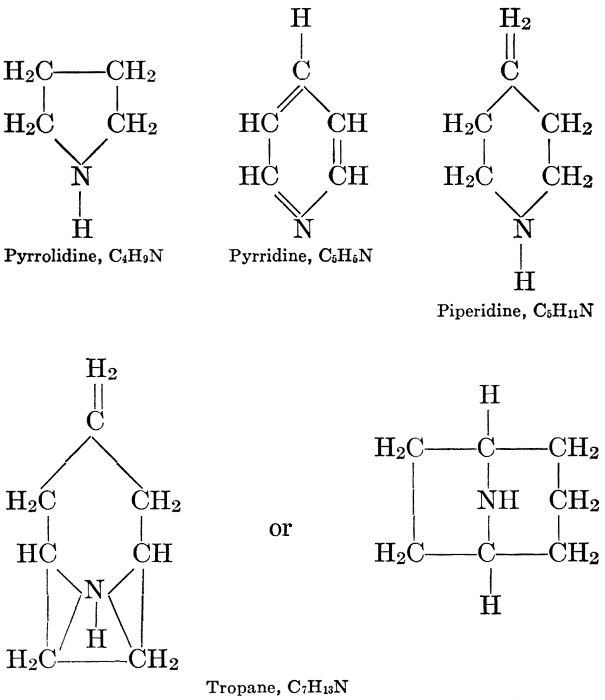
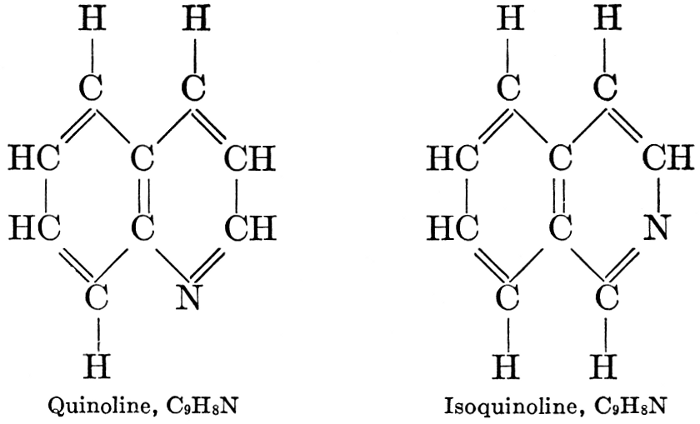
The common alkaloids are distributed in the several groups as follows:
- Pyrridine—piperidine group; piperine, coniine, nicotine.
- Pyrrolidine group; hygrine and stachydrine.
- Tropane group; atropine, hyoscine, cocaine, lupinine.
- Quinoline group; quinine, cinchonine, strychnine, brucine.
- Isoquinoline group; papaverine, hydrastine, morphine, codeine, berberine.
The composition and properties of the individual alkaloids have been extensively studied, because of their medicinal uses. As they have no known metabolic use to the plants which elaborate them, it will not be worth while to consider all of these investigations in detail here. The following facts with reference to certain typical members of each group will serve to illustrate the general constitution and properties of the alkaloids.
Piperine, C17H19O3, is found in black peppers. Its constitution is represented by the following formula, the group which is united to the piperidine ring, in this case, being piperic acid:
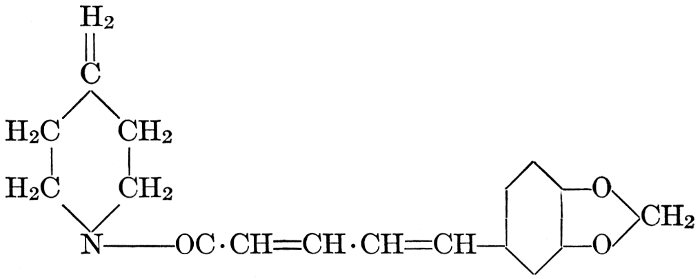
Coniine, C8H17N, is found in the umbelliferous plant, Conium maculatum. Structurally, it is a propyl-piperidine, represented by the following formula:
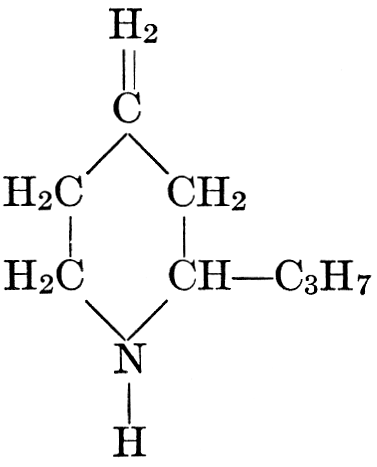
Nicotine, C10H14N2, is the alkaloid of tobacco leaves. It is an extremely poisonous, oily liquid, with a strong odor and a burning taste. Its structural formula shows it to contain both a pyrridine ring and a pyrrolidine ring, linked together thus
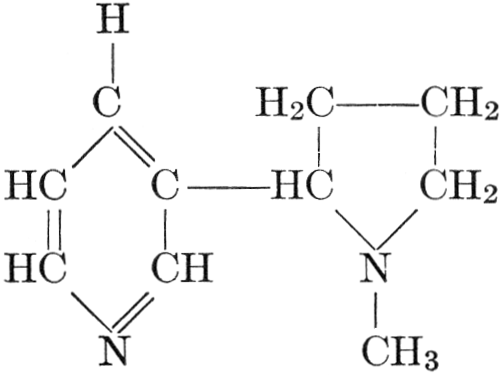
Hygrine, C7H13NO, from coca leaves, is an acetic acid salt of pyrrolidine, represented by the following formula:
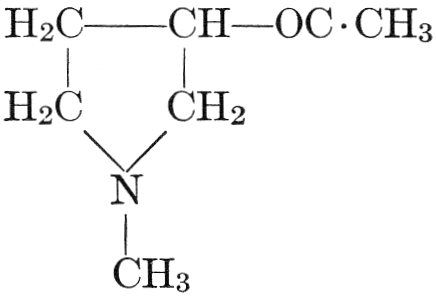
Atropine and hyoscyamine, C17H23NO3, are optical isomers. Atropine is an extremely poisonous, white crystalline compound, which is obtained from deadly nightshade and henbane, and used in medicine, in minute doses, as an agent for reducing temperature in acute cases of fevers. Structurally, it is a tropic acid ester of tropane, represented by the following formula:[Pg 157]
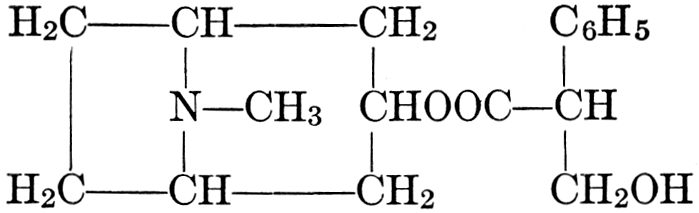
Cocaine, C17H21NO4, is found in coca leaves. It is a white crystalline solid, which is largely used as a local anæsthetic for minor surgical operations. Its structural formula is
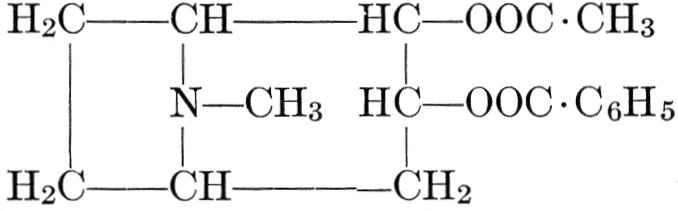
It is, therefore, a di-ester of acetic and benzoic acids with tropane.
Cinchonine, C19H22N2O, and quinine, C20H24N2O2, are alkaloids found in cinchona bark. They are white crystalline solids, which are extensively used in medicine. They have been shown to contain a quinoline group combined with modified piperidine groups, as represented in the following formulas:
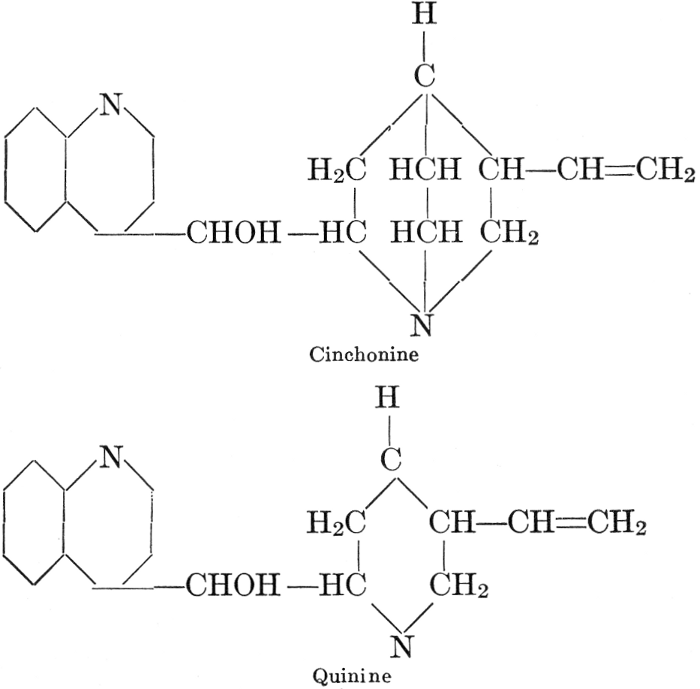
Strychnine, C21H22N2O2, brucine, C21H20(OCH3)N2O2, and curarine are three alkaloids which are present in the seeds of several[Pg 158] species of Strychnos. They are all highly poisonous. Beyond the fact that when they are hydrolyzed they yield quinoline and indole, their composition is unknown.
Morphine, C17H19NO3, is the chief alkaloid of opium, which is the dried juice of young pods of the poppy. Both the alcoholic solution of opium (known as "laudanum") and morphine itself are extensively used in medicine as narcotics to deaden pain. Morphine has an exceedingly complex structure, being a combination of an isoquinoline and a phenanthrene nucleus, which is probably correctly represented by the following formula:
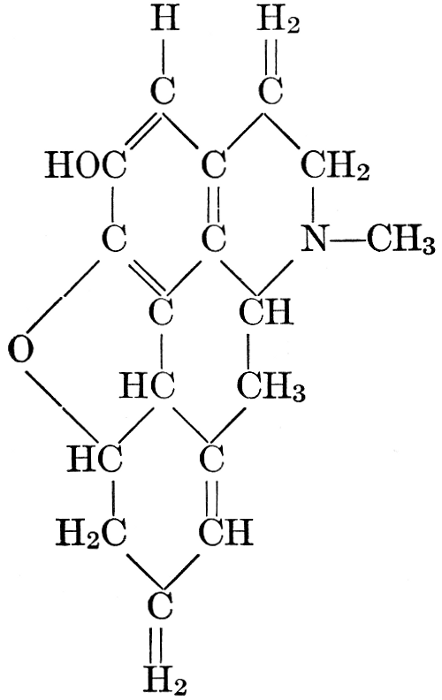
Codeine, C17H18(OCH3)NO2, which is also found in opium, is a methyl derivative of morphine. Papaverine, laudanosine, narcotine, and narceine are four other alkaloids found in opium. They each contain an isoquinoline nucleus, combined by one bond to a benzene ring, with one or more methyl groups and three or more methoxy (OCH3) groups attached at various points around the three characteristic rings. The following formula for laudanosine will illustrate their structure:
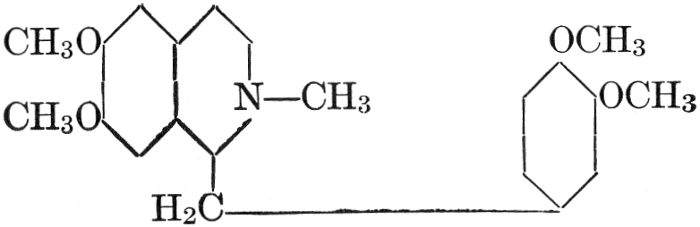
The above discussions of the composition of typical alkaloids clearly indicate the extreme complexity of their molecular structure. It is generally supposed that they are formed by the decomposition of proteins. But they are developed in only a few particular species of plants and are always present in these plants in fairly constant quantities. Hence, it appears that, in these species, the production of alkaloids is in some way definitely connected with protein metabolism; but it is certain that this is not a common relationship, as it is manifested by such a limited number of species of plants, and there is absolutely no knowledge as to its character and functions. Some authorities prefer to regard the alkaloids as waste-products of protein metabolism; but here, again, it is difficult to understand why such products should result in certain species of plants and not in others.
THE PURINE BASES
This is a group of compounds, widely distributed in both plant and animal tissues, all of which are derivatives of the compound known as purine, C5H4N4. All of the naturally occurring compounds of this group may be regarded as derived from purine, either by the addition of oxygen atoms, or by the replacing of one or more of its hydrogen atoms with a methyl (CH3) group or an amino (NH2) group. The following structural formula represents the arrangement of the purine nucleus, the numbers being used to designate the nitrogen or carbon atoms to which the additional atoms, or groups, are attached in the more complex compounds of the group. In purine itself, the four hydrogen atoms are attached in the 2, 6, 7, and 8 positions.
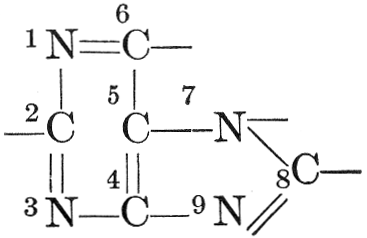
The double bonds, in each case except those between the 4 and 5 carbon atoms, are easily broken apart and readjusted, so that other atoms or groups can be attached to any atom in the nucleus except the 4 and 5 carbon atoms. In all of the statements with reference to the structure of the purine bases, the term "oxy" is used[Pg 160] to mean an oxygen atom attached by both its bonds to one of the carbons in the nucleus, instead of its customary use to mean the monovalent OH group replacing a hydrogen, as in the case of all other nomenclature of organic compounds. With this understanding, reference to the numbered nucleus formula above will make plain the structure of all of the purine bases which are included in the following list:
- Hypoxanthine, C5H4N4O, = 6-monoxypurine.
- Xanthine, C5H4N4O2, = 2,6-dioxypurine.
- Uric acid, C5H4N4O3, = 2,6,8-trioxypurine.
- Adenine, C5H3N4NH2, = 6-aminopurine.
- Guanine, C5H3N4ONH2, = 2-amino-6-oxypurine.
- Theobromine, C5H2N4O2(CH3)2 = 3,7-dimethyl-2,6-dioxypurine, or dimethyl xanthine.
- Theophylline, C5H2N4O2(CH3)2 = 1,3-dimethyl-2,6-dioxypurine.
- Caffeine, C5HN4O2(CH3)3 = 1,3,7-trimethyl-2,6-dioxypurine, or trimethyl xanthine.
In order to make these structural relationships quite clear, the following formulas for uric acid and for caffeine are presented as typical examples:
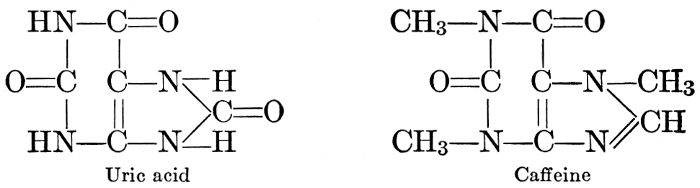
Uric acid is found in the excrement of all animals; in the urine of mammals, and in the solid excrement of birds and reptiles. It is not known to occur in plants.
Xanthine and hypoxanthine occur in animal urine, and also in the tissues of both plants and animals.
Adenine and guanine are constituents of all nucleic acids (see below) and, hence, are found in all plant and animal tissues. Guanine is the chief constituent of the excrement of spiders, and is found also in Peruvian guano. It is also a constituent of the scales of fishes.
Caffeine, theophylline, and theobromine are not found in animal tissues, but are fairly widely distributed in plants. Caffeine and theobromine are the active constituents of tea leaves and coffee[Pg 161] seeds and are found also in cacao beans and kola nuts. The use of these three compounds in the metabolism of the plants which elaborate them is wholly unknown. They are not so directly related to protein metabolism as are the other purine bases.
The purine bases, other than the three mentioned in the preceding paragraph, are undoubtedly intermediate products in protein metabolism. In animals, they constitute a large proportion of the waste-products from the use of proteins in the body. It is not clear that there are similar waste-products in plant metabolism, however. In both plants and animals, the purine bases which are a part of the nucleic acids undoubtedly play an important and essential part in growth, since they form the major proportion of the nucleus, from which all cell-division proceeds.
THE PYRIMIDINE BASES
These compounds do not occur free in plants; but since they are constituent groups in the plant nucleic acids (see below), a brief explanation of their composition is desirable. They are nitrogenous bases, similar to, but somewhat simpler than, the purine bases. Their general composition and structural relationships are illustrated by the following typical formulas:
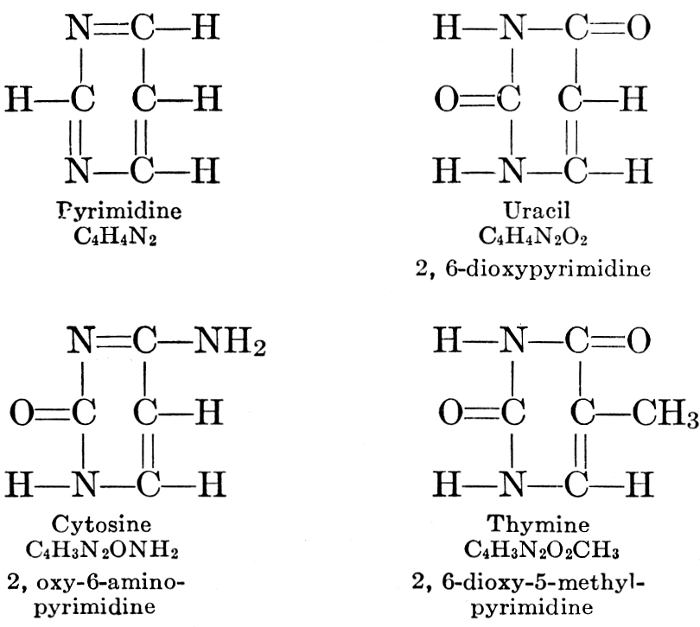
THE NUCLEIC ACIDS
The nuclei of cells are composed almost wholly of complex organic salts, in which proteins constitute the basic part and nucleic acids the acid part. These salts, or esters, are known under the general name "nucleoproteins." The composition of the proteins is discussed in detail in the following chapter, and it seems desirable to present a brief discussion of the constitution of the nucleic acids here; although they are essentially acids rather than vegetable bases.
The nucleic acids are complex compounds consisting of a carbohydrate, phosphoric acid, two purine bases, and two pyrimidine bases. So far as is known, all animal nucleic acids are identical and all plant nucleic acids are identical; but those of plant origin differ from those found in animal cells in the character of the carbohydrate and that of one of the pyrimidine bases which are present in the molecule, as shown in the following tabulation of their composition:
| Animal nucleic acid | Plant nucleic acid |
| Phosphoric acid | Phosphoric acid |
| Hexose (levulose) | Pentose (d-ribose) |
| Guanine | Guanine |
| Adenine | Adenine |
| Cytosine | Cytosine |
| Thymine | Uracil |
The structure of the plant nucleic acid may be represented by the following formula:
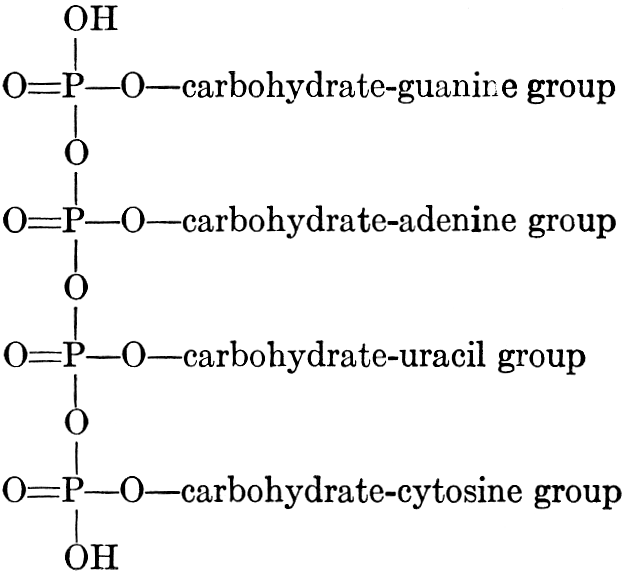
That this is probably a correct representation of the general arrangement in this compound, is indicated by the fact that by different methods of hydrolysis it is possible to split off either the purine and pyrimidine bases, leaving a carbohydrate ester of phosphoric acid; or the phosphoric acid, leaving carbohydrate combinations with the nitrogenous bases.
Nucleic acid, prepared from animal glands which contain large proportions of it, is a white powder, which is insoluble in water, but when moistened forms a slimy mass. It is almost insoluble in alcohol, but dissolves readily in alkaline solutions, forming a colloidal solution which readily gelatinizes (see Chapter on Colloids). Solutions of nucleic acids are optically active, probably because of the carbohydrate constituents.
From their structure and properties, it is apparent that nucleic acids are on the border line between carbohydrates, plant amines, and proteins. They undoubtedly play an important part, both in cell-growth and in the synthesis of proteins from carbohydrates and ammonium compounds.
References
Barger, Geo.—"The Simpler Natural Bases," 215 pages, Monographs on Biochemistry, London, 1914.
Fischer, E.—"Untersuchungen in der Puringruppe, 1882-1906," 608 pages, Berlin, 1907.
Henry, T. A.—"The Plant Alkaloids," 466 pages, Philadelphia, 1913.
Jones, W.—"The Nucleic Acids," 118 pages, Monographs on Biochemistry, London, 1914.
Pictet, A.—"La Constitution Chimique des Alcaloides Vegetaux," 421 pages, Paris, 1897 (2d ed.).
Vaughan, V. C. and Novy, F. G.—"Ptomaines, Leucomaines, Toxins and Antitoxins," 604 pages, Philadelphia, 1896, (3d ed.).
Winterstein, E. and Trier, G.—"Die Alkaloide," 340 pages, Berlin, 1910.
CHAPTER XIII
PROTEINS
The proteins are the most important group of organic components of plants. They constitute the active material of protoplasm, in which all of the chemical changes which go to make up the vital phenomena take place. Combined with the nucleic acids, they comprise the nucleus of the cell, which is the seat of the power of cell-division and, hence, of the growth of the organism. Germ-cells are composed almost exclusively of protein material. Hence, it is not an over-statement to say that proteins furnish the material in which the vital powers of growth and repair and of reproduction are located. A recognition of their importance is reflected in the use of the name "protein," which comes from a Greek word meaning "pre-eminence," or "of first importance."
In addition to the proteins which constitute the active protoplasm, plants also contain large amounts of reserve, or stored, proteins, especially in the seeds. In the early stages of growth, the proteins are present in largest proportions in the vegetative portions of the plant; but as maturity approaches, a considerable proportion of the protein material is transferred to the seeds.
GENERAL COMPOSITION OF PROTEINS
The plant proteins are fairly uniform in their percentage composition. The analyses of some sixteen different plant proteins show the following maximum limits of percentages of the different chemical elements which they contain: Carbon, 50.72-54.29; hydrogen, 6.80-7.03; nitrogen, 15.84-19.03; oxygen, 20.86-24.29; sulfur, 0.17-1.09. Animal proteins vary more widely, both in percentage composition and in properties, than do those of plant origin.[Pg 165]
Protein molecules are very large and, in the case of the so-called "conjugated proteins" in particular, their structure is very complex. The molecular weight of some of the proteins has been determined directly, in the case of those particular ones which can be prepared in proper form for the usual determination of molecular weight by the osmotic pressure method; and has been computed for various others, from the percentage of sulfur found on analysis, or (in the case of the hæmoglobin of the blood) from the proportion by weight of oxygen absorbed. From these determinations and computations, the following formulas for certain typical proteins have been calculated: for zein (from Indian corn), C736H1161N184O208S3; for gliadin (from wheat), C685H1068N196O211S5; for casein (from milk), C708H1130N180O224S4P4; for egg-albumin, C696H1125N175O220S8. These few examples will serve to illustrate the enormous size and complexity of the protein molecule. The conjugated proteins are still more complex than the simple proteins whose formulas are here presented.
Fortunately for the purposes of the study of the chemistry of the proteins, however, it has been found that most of the common plant proteins, known as the "simple proteins," can easily be hydrolyzed into their constituent unit groups, which are the comparatively simple amino-acids, whose composition and properties are well understood. A study of the results of the hydrolysis of some twenty common plant proteins has shown that it is rarely possible to recover the amino-acids in sufficient quantities to account for a full 100 per cent of the material used, the actual percentage of amino-acids recovered usually totaling from 60 to 80 per cent. The remaining material is supposed to be also composed of amino-acids which are linked together in some arrangement which is not broken apart by any method of hydrolysis which has yet been devised. This view is borne out by the fact that substances which exhibit all the characteristic properties of proteins have been artificially synthetized, by using only amino-acid compounds. Animal proteins often show a much larger proportion of unhydrolyzable material than do plant proteins.[Pg 166]
AMINO-ACIDS AND PEPTID UNITS
The products of hydrolysis of the common simple proteins are all amino-acids. These are ordinary organic acids with one (or more) of the hydrogen atoms of the alkyl group replaced by a —NH2 (or sometimes by a —NH—) group. They may be regarded as ammonia, NH3, with one of its hydrogen atoms replaced by an acid radical; or as the acid with one of its hydrogens replaced by the NH2 group. For example, an amino-acid derived from acetic acid, CH3·COOH, is glycine, or amino-acetic acid, CH2NH2·COOH; from propionic acid, CH3·CH2·COOH, there may be obtained either α-amino-propionic acid, CH3·CHNH2·COOH, or β-amino-propionic acid, CH2NH2·CH2·COOH, etc.
All of the amino-acids which result from the hydrolysis of proteins are α-amino-acids, that is to say, the NH2 group is attached to the α-carbon atom, i.e., the one nearest to the COOH group. Hence, the general formula for all the amino-acids which are found in plants is R·CHNH2·COOH.
These amino-acids contain both the basic NH2 group and the acid COOH group. For this reason, they very easily unite together, in the same way that all acids and bases unite, to form larger molecules, the linkage taking place between the basic NH2 group of one molecule and the acid COOH group of the other, as indicated by the following equation:
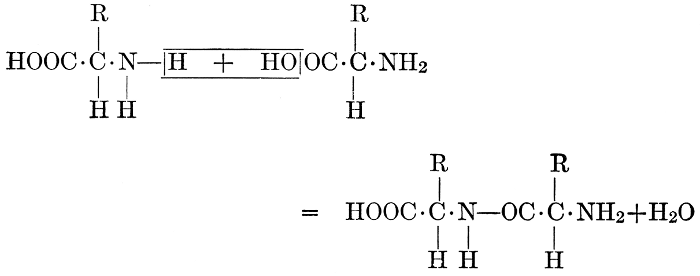
It is obvious that the compound thus formed still contains a free NH2 group and a free COOH group, and is, therefore, capable of linking to another amino-acid molecule in exactly the same way; and so on indefinitely. In actual laboratory experiments, as[Pg 167] many as eighteen of these amino-acid units have been caused to unite together in this way, and the resulting compounds thus artificially prepared have been found to possess the characteristic properties of natural proteins.
These artificially prepared, protein-like, substances have been called "polypeptides," and the individual amino-acids which unite together to form them are called "peptides." Thus, a compound which contains three such units linked together is called a "tripeptid"; one which contains four, a "tetrapeptid." The use of the term "peptid" was suggested by the fact that these amino-acids are produced from the hydrolysis of proteins by the digestive enzyme pepsin.
The peptid units of any such complex as those which have been referred to in the preceding paragraphs may be linked together in a great variety of ways. Thus, in a tetrapeptid containing units which may be designated by the letters a, b, c, and d, the arrangement may be in the orders abcd, bacd, acbd, dbca, etc., etc. Similarly, the same peptid unit may appear in the molecule in two or more different places. Hence, the number of possible combinations of amino-acids into protein molecules is very great. Further, it is possible that the peptid units in natural proteins may be united together through other linkages than the one illustrated above, as they often contain alcoholic OH groups in addition to the basic NH2 groups, and these OH groups may form ester-linkages with the acid (COOH) groups of other units. Still other acid and basic groups are present in some of the amino-acids which have been found in natural proteins, so that the possibility of variation in the polypeptid linkages is almost limitless.
INDIVIDUAL AMINO-ACIDS FROM PROTEINS
About twenty different amino-acids have been isolated from the products of hydrolysis of natural proteins, and this number is being added to from time to time, as the methods of isolation and identification of these compounds are improved. Many of these same amino-acids have been found in free form in plant tissues, particularly in rapidly growing buds, or shoots, or in germinating seeds, where they undoubtedly exist as intermediate products in the transformation of proteins into other types of compounds.[Pg 168]
These amino-acids, grouped according to the characteristic groups which they contain, are as follows:
- Monoamino-monocarboxylic acids:
- Glycine, C2H5NO2, CH2NH2·COOH, amino-acetic acid.
- Alanine, C3H7NO2, CH3·CHNH2·COOH, amino-propionic acid.
- Serine, C3H7NO3, CH2OH·CHNH2·COOH, oxy-amino-propionic acid.
- Valine, C5H11NO2,
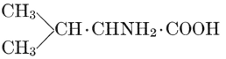 , amino-isovalerianic acid.
, amino-isovalerianic acid.
- Leucine, C6H13NO2,
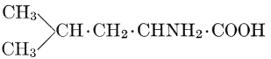 , amino-isocaproic acid.
, amino-isocaproic acid.
- Isoleucine, C6H13NO2,
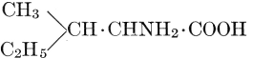 , amino-methylethyl-propionic acid.
, amino-methylethyl-propionic acid.
- Phenylalanine, C9H11NO2,
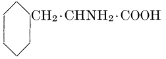 , phenyl-amino-propionic acid.
, phenyl-amino-propionic acid.
- Tyrosine, C9H11NO3,
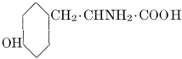 , paraoxy-phenylalanine.
, paraoxy-phenylalanine.
- Cystine, C6H12N2O4S2, HOOC·CHNH2·CH2S—SH2C·CHNH2·COOH, di(thio-amino-propionic acid).
- Monoamino-dicarboxylic acids:
- Aspartic acid, C4H7NO4, HOOC·CH2·CHNH2·COOH, mino-succinic acid.
- Glutamic acid, C5H9NO4, HOOC·CH2·CH2·CHNH2·COOH, amino-glutaric acid.
-
Diamino-monocarboxylic acids:
- Ornithine, C5H12N2O2, H2N·CH2·CH2·CH2·CHNH2·COOH, di-amino-valerianic acid.
- Lysine, C6H14N2O2, H2N·CH2·CH2·CH2·CH2·CHNH2·COOH, di-amino-caproic acid.
- Arginine, C6H14N4O2,
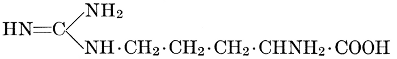 , guanidine-amino-valerianic acid.
, guanidine-amino-valerianic acid.
- Di-amino-oxysebacic acid, C11H12N2O3.
- Di-amino-trioxydodecanic acid, C12H26N2O3.
- Monoimido-monocarboxylic acids:
- Proline, C5H9NO2,
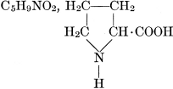 , pyrrolidine-carboxylic acid.
, pyrrolidine-carboxylic acid.
- Oxyproline, C5H9NO3, proline with one (OH) group.
- Proline, C5H9NO2,
- Monoimido-monoamino-monocarboxylic acids:
- Histidine, C6H9N3O2,
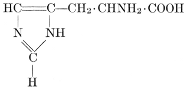 , imidazole-amino-propionic acid.
, imidazole-amino-propionic acid.
- Tryptophane, C11H12N2O2,
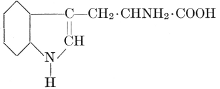 , indole-amino-propionic acid.
, indole-amino-propionic acid.
- Histidine, C6H9N3O2,
As has been said, other amino-acids are being found, from time to time, as additional proteins are examined, or as better methods of examination of the cleavage products of the natural proteins are devised.[Pg 170]
COMPOSITION OF PLANT PROTEINS
The distribution of the different amino-acids in some of the different plant proteins which have been examined in this way is shown in the following table:
| Gliadin (whaet). |
Hordein (barley). |
Zein (corn). |
Legumin (vetch). |
Edestin (hemp). |
Globulin (squash seed). |
Amandin (almonds). |
|
|---|---|---|---|---|---|---|---|
| 71.46 | 78.16 | 85.27 | 62.65 | 82.38 | 55.77 | 59.00 | |
| Glycine | 0.02 | 0.00 | 0.00 | 0.39 | 3.80 | 0.57 | 0.51 |
| Alanine | 2.00 | 0.43 | 9.79 | 1.15 | 3.60 | 1.92 | 1.40 |
| Valine | 0.21 | 0.13 | 1.88 | 1.36 | 6.20 | 0.26 | 0.16 |
| Leicine | 5.61 | 5.67 | 19.55 | 8.80 | 14.50 | 7.32 | 4.45 |
| Proline | 7.06 | 13.73 | 9.04 | 4.04 | 4.10 | 2.82 | 2.44 |
| Phenylalanine | 2.35 | 5.03 | 6.55 | 2.87 | 3.09 | 3.32 | 2.53 |
| Aspartic acid | 0.58 | ..... | 1.71 | 3.21 | 4.50 | 3.30 | 5.42 |
| Glutamic acid | 42.98 | 43.19 | 26.17 | 18.30 | 18.84 | 12.35 | 23.14 |
| Serine | 0.13 | ? | 1.02 | ? | 0.33 | ? | ? |
| Cystine | 0.45 | ? | ? | ? | 1.00 | 0.23 | ? |
| Tyrosine | 1.20 | 1.67 | 3.55 | 2.42 | 2.13 | 3.07 | 1.12 |
| Arginine | 3.16 | 2.16 | 1.55 | 11.06 | 14.17 | 14.44 | 11.85 |
| Histidine | 0.61 | 1.28 | 0.43 | 2.94 | 2.19 | 2.63 | 1.58 |
| Lysine | ..... | ..... | ..... | 3.99 | 1.65 | 1.99 | 0.70 |
| Tryptophane | present | present | present | present | present | present | present |
| Ammonia | 5.11 | 4.87 | 3.64 | 2.12 | 2.28 | 1.55 | 3.70 |
At the time when these analyses were made, a method for the quantitative estimation of tryptophane had not been devised, although one is now available. The addition of the percentages of tryptophane and of other amino-acids for which methods of determination are not yet known, would bring the total, in each case, more nearly up to the full 100 per cent. These data will serve to show how widely the different plant proteins vary in the proportions of the different amino-acids which they contain. Animal proteins have been found to be still more variable in composition.
In the use of the proteins as food for animals, it appears that the different amino-acids are in some way connected with the[Pg 171] different physiological functions which the proteins have to perform in the animal body: thus, tryptophane is absolutely essential to the maintenance of life, but does not promote growth; lysine, on the other hand, definitely promotes growth, so that animals which have been maintained without any increase in weight for many months immediately begin to grow when furnished with a diet in which lysine is a constituent; while arginine seems to be definitely associated with the reproductive function; and cystine, with the growth of hair, feathers, etc. It is not known whether there is any similar relation of amino-acids to the functions of different proteins in plant metabolism.
The separation of the individual amino-acids from the mixture which results from the hydrolysis of any given protein is a long and tedious process and, at best, yields only moderately satisfactory results. For that reason, it has recently been almost entirely abandoned in favor of the separation devised by Van Slyke, which divides the total nitrogenous matter in the mixture resulting from the hydrolysis of a protein into the following groups; ammonia N, humin (or melanin) N, cystine N, arginine N, histidine N, lysine N, amino N of the filtrate, and non-amino N of the filtrate. These groups can be conveniently and fairly accurately separated out of the hydrolysis mixture, by means of various precipitating agents, and the quantity of N in the several precipitates determined by the usual Kjeldahl method. The actual process for these separations need not be discussed here, as it is given in detail in all standard text-books dealing with the methods of biochemical analysis. The distribution of the nitrogen in any given protein into these various groups is characteristic for that particular protein, and the process serves both as a means of identification of individual proteins and a method for tracing their changes through various vital, or biochemical, transformations.
GENERAL PROPERTIES OF THE PROTEINS
Individual proteins differ slightly in their characteristics, but in general they are all alike in the following physical and chemical properties.[5][Pg 172]
Physical Properties.—(1) The proteins are all colloidal in character, that is, they form solutions in water, out of which they cannot be dialyzed through parchment, or other similar membranes. (2) All natural proteins, when in colloidal solution, may be coagulated, forming a semi-solid gel, which cannot again be rendered soluble except by decomposition. The most familiar example of this type of coagulation is that of egg-albumin, when eggs are cooked. This coagulation may be produced by heat, by the action of certain enzymes, or by the addition of alcohol to the solution. (3) All solutions of plant proteins are optically active, rotating the plane of polarized light to the left, in every case. (4) Proteins are precipitated out of their solutions, without change in the composition of the protein, by saturating the solution with various neutral salts of the alkali, or alkaline earth, metals, such as sodium chloride, ammonium sulfate, magnesium sulfate, etc. This is only another way of saying that the proteins are insoluble in strong salt solutions. Separation from solution by the addition of salts is different from coagulation by heat, etc., as in this case simple dilution of the salt solution will cause the protein to redissolve, whereas a coagulated protein cannot be redissolved without some change in its composition.
Chemical Properties. (1) Precipitation reactions.—The proteins have both acid and basic properties (due to the presence in their molecules of both free NH2 groups and free COOH groups). Bodies of this kind are known as "amphoteric electrolytes," since they yield both positive and negative ions, if dissociated. The proteins readily form salts, which are generally insoluble in water, with strong acids. For this reason, they are generally precipitated out of solution by the addition of the common mineral acids. They are also precipitated by many of the "alkaloidal reagents," to which reference has been made in the preceding chapter, namely, phosphotungstic, phosphomolybdic, tannic, picric, ferrocyanic, and trichloracetic acids, the double iodide of potassium, mercuric iodide, etc. The precipitates produced by strong mineral acids are often soluble in excess of the acid, with the formation of certain so-called "derived proteins," which are probably products of the partial hydrolysis of the protein.[Pg 173]
The proteins are also precipitated out of solution by the addition of small amounts of salts of various heavy metals, such as the chlorides, sulfates, and acetates of iron, copper, mercury, lead, etc. This precipitation is different than that caused by the saturation of the solution with the salts of the alkali metals, as in this case the metal unites with the protein to form definite, insoluble salts, which cannot be redissolved except by treatment with some reagent which removes the metal from its combination with the protein (hydrogen sulfide is commonly used for this purpose).
(2) Color reactions.—Certain specific groups which are present in most proteins give definite color reactions with various reagents. It is apparent that any individual protein will respond to a particular color reaction, or will not do so, depending upon whether the particular group which is responsible for the color in question is present in that particular protein. Color reactions to which most of the common plant proteins respond are the following ones:
(a) Biuret Reaction.—Solutions of copper sulfate, added to an alkaline solution of a protein, give a bluish-violet color if the substance contains two, or more, —CONH— groups united together through carbon, nitrogen, or sulfur atoms. Inasmuch as most natural proteins contain several such groups, the biuret reaction is a very general test for proteins.
(b) Millon's Reaction.—A solution of mercuric nitrate containing some free nitrous acid (Millon's reagent) produces a precipitate which turns pink or red, whenever it is added to a solution which contains tyrosine, or a tyrosine-containing protein.
(c) Xanthoproteic Acid Reaction.—This is the familiar yellow coloration which is produced whenever nitric acid comes in contact with animal flesh. It is caused by the action of nitric acid on tyrosine. The color is intensified by heating, and is changed to orange-red by the addition of ammonia.
(d) Adamkiewicz's Reaction.—If concentrated sulfuric acid be added to a solution of a protein to which some acetic acid (or[Pg 174] better, glyoxylic acid) has previously been added, a violet color is produced. This color will appear as a ring at the juncture of the two liquids, if the sulfuric acid is poured carefully down the sides of the tube, or throughout the mixture if it is shaken up. It depends upon the interaction of the glyoxylic acid (which is generally present as an impurity in acetic acid) upon the tryptophane group, and is therefore given by all proteins which contain tryptophane.
(e) Molisch's reaction for furfural will be shown by those proteins which contain a carbohydrate group. In applying this test, the solution to be tested is first treated with a few drops of an alcoholic solution of α-naphthol, and then concentrated sulfuric acid is poured carefully down the sides of the test-tube. If carbohydrates are present, either free or as a part of a protein molecule, a red-violet ring forms at the juncture of the two liquids.
(f) Sulfur Test.—If a drop of a solution of lead acetate be added to a solution containing a protein, followed by sufficient sodium hydroxide solution to dissolve the precipitate which forms, and the mixture is heated to boiling, a black or brown coloration will be produced if the protein contains cystine, the sulfur-containing amino-acid.
FOOTNOTES:
[5] Since the proteins are essentially colloidal in nature, many of the terms used in the discussions of their properties, and these properties themselves, will be better understood after the chapter dealing with the colloidal condition of matter has been studied. A more logical arrangement so far as the systematic study of these properties is concerned would be to take up chapter XV before undertaking the study of the proteins (this order is actually followed in some texts on Physiological Chemistry). But from the standpoint of the consideration of the various groups of organic components of plants, it seems a better arrangement to consider these groups in sequence, and then to discuss the various physical-chemical phenomena which govern their activity. However, it is recommended that the student refer at once to Chapter XV for an explanation of any terms used here, which may not be familiar to him; and that after the study of Chapter XV, he return to this chapter dealing with the proteins for an illustrative study of the applications of the principles presented there.
THE CLASSIFICATION OF THE PROTEINS
Formerly, the classification of proteins was based almost wholly upon their solubility and coagulation reactions. More recently, since their products of hydrolysis have been extensively studied, their classification has been modified, in attempts to make it correspond as closely as possible to their chemical constitution and physical properties. As knowledge of these matters progresses, the schemes of classification change. On that account, no one definite scheme is universally used. For example, the English system varies considerably from the one commonly used by American biochemists, which is the one presented below.
The proteins are divided into three main classes, as follows:
- Simple proteins, which yield only amino-acids when hydrolyzed.
- Conjugated proteins, compounds of proteins with some other non-protein group.
- Derived proteins, decomposition products of simple proteins.
The first two of these classes comprise all the natural proteins; while the third includes the artificial polypeptides and proteins which have been modified by reagents.
These major classes are further subdivided into the following sub-classes, which depend in part upon the solubilities of the individual proteins, and in part upon the nature of their products of hydrolysis:
- The Simple Proteins
- Albumins—soluble in water and dilute salt solutions, coagulated by heat.
- Globulins—insoluble in water, soluble in dilute salt solutions, coagulated by heat.
- Glutelins—insoluble in water or dilute salt solutions, soluble in dilute acids or alkalies, coagulated by heat.
- Prolamins—insoluble in water, etc., soluble in 80 per cent alcohol.
- Histones—soluble in water, insoluble in ammonia, not coagulated by heat.
- Protamines—soluble in water and ammonia, not coagulated by heat, yielding large proportions of diamino-acids on hydrolysis.
- Albuminoids—insoluble in water, salt solutions, acids, or alkalies.
- Conjugated Proteins
- Chromoproteins—compounds of proteins with pigments.
- Glucoproteins—compounds of proteins with carbohydrates.
- Phosphoproteins—proteins of the cytoplasm, containing phosphoric acid.
- Nucleoproteins—proteins of the nucleus, containing nucleic acids.
- Lecithoproteins—compounds of proteins with phospholipins.
- Lipoproteins—compounds of proteins with fats, existence in nature doubtful, artificial forms easily prepared.
- Derived Proteins
- Primary protein derivatives.
- Proteans—first products of hydrolysis, insoluble in water.
- Metaproteins—result from further action of acids or alkalies, soluble in weak acids and alkalies, but insoluble in dilute salt solutions.
- Coagulated proteins—insoluble forms produced by the action of heat or alcohol.
- Secondary protein derivatives.
- Proteoses—products of hydrolysis, soluble in water, not coagulated by heat, precipitated by saturation of solution with ammonium sulfate.
- Peptones—products of further hydrolysis soluble in water, not coagulated by heat, not precipitated by ammonium sulfate, give biuret reaction.
- Peptides—individual amino-acids, or poly-peptides, may or may not give biuret reaction.
- Primary protein derivatives.
The plant proteins which have been investigated, thus far, fall into these groups as follows:
| 1A. | Albumins | |||
| Leucosin, | found in the | seeds of | wheat, rye and barley. | |
| Legumelin, | " | " | pea, horse-bean, vetch, soy-bean, lentil, cowpea, adzuki-bean. | |
| Phaselin, | " | " | kidney-bean. | |
| Ricin, | " | " | castor-bean. | |
| 1B. | Globulins | |||
| Legumin, | found in the | seeds of | pea, horse-bean, lentil and vetch. | |
| Vignin, | " | " | cowpea. | |
| Glycinin, | " | " | soy-bean. | |
| Phaseolin, | " | " | beans (Phaseolus spp.) | |
| Conglutin, | " | " | lupines. | |
| Vicilin, | " | " | pea, horse-bean, lentil. | |
| Corylin, | " | " | hazel nut. | |
| Amandin, | " | nuts of | almond and peach. | |
| Juglansin, | " | seeds of | walnut and butternut. | |
| Excelsin, | " | " | Brazil nut | |
| Edestin, | " | hemp seed. | ||
| Avenalin, | " | oats. | ||
| Maysin, | " | corn. | ||
| Castanin, | " | seeds of European chestnut. | ||
| And, crystalline globulins found in the seeds of flax, squash, castor-bean, sesame, cotton, sunflower, radish, rape, mustard, and in cocoanuts, candlenuts, and peanuts. |
||||
| 1C. | Glutelins | |||
| Glutenin, | found in the | seeds of | wheat. | |
| Oryzenin, | " | " | rice. | |
| 1D. | Prolamins | |||
| Gliadin, | found in the | seeds of | rye, wheat, with glutenin forms "gluten." | |
| Hordein, | " | " | barley. | |
| Zein, | " | " | corn. | |
1E-1G. Histones, Protamines and Albuminoids.—So far as is now known, no representatives of these classes are found in plants.
2. Conjugated Proteins.—There is no conclusive evidence of the existence in plants of any of the conjugated proteins, other[Pg 177] than the nucleoproteins and the chromoproteins, the composition and properties of which have been discussed in previous chapters. The nucleoproteins undoubtedly occur in the embryos of many, if not all, seeds.
3. Derived Proteins.—Representatives of the various types of derived proteins are undoubtedly found as temporary intermediate products in plants, both as products of hydrolysis produced during the germination of seeds and as intermediate forms in the synthesis of proteins. So far as is known, however, they do not occur as permanent forms in any plant tissues. They have been prepared in large numbers and quantities, by the hydrolysis of the natural proteins and the artificial synthesis of polypeptides.
In the present state of our knowledge concerning the functioning of the proteins, no significance in the physiology of plant life, or metabolism, is to be attached to the particular type of protein material which it contains, at least so far as the simple proteins of the cytoplasm are concerned.
DIFFERENCES BETWEEN PLANT AND ANIMAL PROTEINS
A much larger variety of protein materials is found in animal tissues than in plants. This is undoubtedly because different animal organs perform so much more varied physiological functions than do those of plants. Three groups of simple proteins, the histones, the protamines, and the albuminoids, which are quite common in animal tissues, are entirely unknown in plants. Further, conjugated proteins of greater complexity and more varied structure are found in animal tissues, especially in the brain, nerve-cells, etc., than in plants.
Plant proteins, in general, usually contain larger proportions of proline and of glutamic acid than are found in animal proteins; also more arginine than is found in any of the animal proteins except the protamines, which contain as high as 85 per cent of this amino-acid.
Of the twenty-five plant proteins which have thus far been hydrolyzed and studied from this standpoint, all contained leucine, proline, phenylalanine, aspartic acid, glutamic acid, tyrosine, histidine, and arginine; two gave no glycine; two others, no alanine; four contained no lysine; and one, no tryptophane. Zein, the principal protein of corn contains no glycine, lysine, or[Pg 178] tryptophane. It is not sufficient to support animal life and promote growth, if used as an exclusive source for protein for food.
THE EXTRACTION OF PROTEINS FROM PLANT TISSUES
Since proteins are indiffusible, it is essential that the cell-walls of the tissue shall be thoroughly ruptured as the first step in any process for the extraction of these compounds from plant tissues. This is usually accomplished by grinding the material as finely as possible, preferably with the addition of sharp quartz sand, or broken glass, to aid in the tearing of the cell-wall material.
The solvent to be used in extracting the proteins from this finely ground material depends upon the nature and solubility of the proteins which are present, and also upon whether it is desired to separate the proteins which may be present in the plant, during the process of the extraction. A glance at the scheme of classification of the proteins will show the following solubilities which serve as a guide to the procedure to be followed: (a) proteoses, albumins, and some globulins may be extracted with water; (b) globulins and most of the water-soluble proteins may be extracted by using a 10 per cent solution of common salt; (c) prolamines are extracted by 70-90 per cent alcohol; glutelins and prolamins dissolve in dilute acids or dilute alkali.
A common procedure is to extract groups (a) and (b), using a 10 per cent salt solution as the solvent, and then to separate the albumins, globulins, etc., from this solution by suitable precipitants; then to treat the material with 80 per cent alcohol, to extract the prolamines; and finally with dilute alkali, to extract the glutelins. The dissolved proteins in each extract can be subsequently purified by dialysis, precipitation, etc. The insoluble proteins can be studied only after removing the other materials associated with them in the tissue, by suitable mechanical or chemical means.
THE SYNTHESIS OF PROTEINS IN PLANTS
The synthesis of proteins in plants is not a process of photosynthesis, as it can take place in the dark and in the absence of chlorophyll, or any other energy-absorbing pigment. However, protein-formation normally takes place in conjunction with car[Pg 179]bohydrate-formation. The carbon, hydrogen, and oxygen necessary for protein synthesis are undoubtedly obtained from carbohydrates. The nitrogen and sulfur come from the salts absorbed from the soil through the roots and brought to the active cells in the sap. Atmospheric nitrogen cannot be used by plants for this purpose, except in the case of certain bacteria and other low plants, notably the bacteria which live in symbiosis with the legumes in the nodules on the roots of the host plants. In general, the sulfur must come in the form of sulfates and the nitrogen in the form of nitrates; although many plants can make use of ammonia for protein-formation. Presumably, the nitrate nitrogen must be reduced in the plant to nitrites, and then to ammonia form, in order to enter the amino-arrangement required for the greater proportion of the protein nitrogen.
The mechanism by which ammonia nitrogen becomes amino-acids in the plant is not understood. Artificial syntheses of amino-acids, by the action of ammonia upon glyoxylic acid and sorbic acid, both of which occur in plants and may be obtained by the oxidation of simple sugars, have been accomplished, and it seems probable that similar reactions in the plant protoplasm may give rise to the various amino-acids which unite together to form proteins. Nothing is known, however, of the process by which the more complicated closed-ring amino-acid compounds, such as proline, histidine, or tryptophane, are synthetized.
The condensation of amino-acids into proteins, or the reverse decomposition, is very readily accomplished in all living protoplasm, under the influence of special protein-attacking enzymes, which are almost universally present in the cytoplasm. These reactions in connection with the proteins are similar to the easy transformation of sugars to starches, and vice versa, under the action of the corresponding carbohydrate-attacking enzymes.
PHYSIOLOGICAL USES OF PROTEINS
There can be no doubt that the all-important rôle of proteins, in either plant or animal tissue, is to furnish the colloidal protoplasmic material in which the vital phenomena take place. Their occurrence in seeds, and other storage organs, is, of course, in order to provide the protoplasm-forming material for the young seedling plant.[Pg 180]
They are, moreover, the source for the material which goes into some of the secretion groups of organic compounds; as they are easily broken down by various agents of decomposition into nitrogen-free alcohols, aldehydes, and acids, which produce the essential oils, pigments, etc.
Much, if not all, of their physiological activity is due to their colloidal nature, the importance and effects of which will be more apparent after the chapters dealing with the colloidal condition of matter and with the physical chemistry of protoplasm have been studied.
References
Abderhalden, E.—"Neuere Ergebnisse auf dem Gebiete der Speziellen Eiweisschemie," 128 pages, Jena, 1909.
Fischer, E.—"Untersuchungen über Aminosäuren, Polypeptide, und Proteine, 1899-1906," 770 pages, Berlin, 1906.
Mann, G.—"Chemistry of the Proteids," 606 pages, London, 1906.
Osborne, T. B.—"The Vegetable Proteins," 138 pages, Monographs on Biochemistry, London, 1909.
Plimmer, R. H. A.—"The Chemical Constitution of the Proteins, Part I, Analysis," 188 pages; and "Part II, Synthesis, etc." 107 pages, Monographs on Biochemistry, London, 1917. (3d ed.).
Robertson, T. B.—"The Physical Chemistry of the Proteins," 477 pages, New York, 1918.
Schryber, S. B.—"The General Characters of the Proteins," 86 pages, Monographs on Biochemistry, London, 1909.
Underhill, F. P.—"The Physiology of the Amino-acids," 169 pages, 13 figs. 1 plate. Yale University Press, 1915.[Pg 181]
CHAPTER XIV
ENZYMES AND THEIR ACTION
The characteristic difference between the reactions of inorganic compounds and those of organic substances lies in the rapidity, or velocity, of the chemical changes involved. Speaking generally chemical reactions take place between substances which are in solution, so that they may come into sufficiently intimate contact that chemical action between them can take place. There are, of course, occasional examples of reactions between dry solids, such as the explosion of gunpowder, etc., but the general rule is that reacting materials must be in either colloidal or true solutions.
Inorganic materials, when dissolved in water, usually ionize very readily. That is, they are not only disintegrated into individual molecules, but a considerable proportion of these molecules separate into their constituent ions. When solutions containing ionized compounds are brought together, conditions for chemical interaction are ideal, and the reaction proceeds with such tremendous rapidity as to be completed almost instantaneously, in most cases.
Organic compounds, on the other hand, ionize only very slowly, if at all. Hence, reactions between organic compounds, even when they are in solution, proceed very slowly unless carried on at high temperatures, under increased pressure, or under the influence of some catalytic agent. Even under the stimulation of these reaction-accelerating agencies, most chemical changes in organic compounds when carried on in the laboratory, require several hours or even days and sometimes weeks, for their completion. But when similar reactions take place in living organisms, they proceed with velocities which resemble those of inorganic compounds in the laboratory. This difference between the velocity of organic reactions when carried on under artificial conditions in the laboratory (often spoken of as "in vitro") as compared with that of the same reactions when they take place in a living[Pg 182] organism ("in vivo"), is due to the universal presence in the living protoplasm of certain organic catalysts, known as enzymes.
ENZYMES AS CATALYSTS
The phenomenon known as "catalysis" is of common occurrence in both inorganic and organic chemistry. The effect of a small amount of manganese dioxide in aiding in the liberation of oxygen from potassium chlorate is an example which is familiar to all students of elementary chemistry. Similarly, spongy platinum accelerates the oxidation of sulfur dioxide to sulfur trioxide, in the commercial manufacture of sulfuric acid. Again, the hydrolysis of sucrose into fructose and glucose proceeds very slowly in the presence of water alone, but if a little hydrochloric acid or sulfuric acid be added to the solution, the velocity of the hydrolysis is enormously accelerated. Many other examples of the accelerating effect of various chemicals upon reactions into which they do not themselves enter, might be cited.
The essential features of all such catalytic actions are: (1) the velocity of the reaction is greatly altered, usually accelerated; (2) the catalytic agent does not appear as one of the initial substances, or end-products, of the reaction, and is not itself altered by the chemical change which is taking place; (3) the accelerating effect is directly proportional to the amount of the catalyst which is present; (4) relatively small amounts of the catalyst produce very large results in the reacting mixture; and (5) the catalysts cannot themselves initiate reactions, but only influence the velocity of reactions which would otherwise take place at a different rate (usually much more slowly) in the absence of any catalytic agent.
Enzymes conform to all of these properties of catalysts, and are commonly defined as the "catalysts of living matter." They are almost universally present in living organs of every kind, and perform exceedingly important functions, both in the building-up of synthetic materials and in the rendering soluble of the food of both plants and animals, so that it can be translocated from place to place through the tissues of the organism.
Enzymes differ from inorganic catalysts in being destroyed by heat, in not always carrying the reaction to the same stage as does the inorganic catalyst which may accelerate the same reaction, and[Pg 183] in producing different changes in the same substance by different enzymes.
The name "enzyme" comes from Greek words meaning "in yeast," as the nature and effect of the enzyme involved in the alcoholic fermentation of sugars by yeast were those which were first recognized and understood. It was at first thought, by Pasteur and his students, that fermentation is the direct result of the life activities of the yeast plant. Later, it was found that water extracts from sprouted barley, from almond seeds, and from the stomach, pancreas, etc., were able to bring about the decomposition of starch, of amygdalin, and of proteins, respectively, in a way which seemed to be quite comparable to the fermentative action of yeasts. Hence, it was thought that there were two varieties of active agents of this kind, one composed of living cells and the other non-living chemical compounds, and these were called the "organized ferments" and the "unorganized ferments," respectively. However, in 1897, Büchner found that by grinding yeast cells with sharp sand until they were completely disintegrated and then submitting the mass to hydraulic pressure, he could obtain a clear liquid, entirely free from living cells, which was just as active in producing fermentation as was the yeast itself. This discovery paved the way for a long series of investigations, which have conclusively demonstrated that there is no distinction between "organized" and "unorganized" ferments, that all living organisms perform their characteristic functions by means of the enzymes which they contain, and that these enzymes can bring about their characteristic catalytic effects outside the cell, or tissue which elaborates them, just as well as within it, provided only that the conditions of temperature, acidity or alkalinity of the medium, etc., are suitable for the particular enzyme action which is under consideration.
GENERAL PROPERTIES OF ENZYMES
Since enzymes are catalysts, it is plain that an accurate description of their activity should, in each case, refer to the influence which they exert upon some definite reaction velocity. But since the phrases necessary to describe such an effect are cumbersome and inconvenient, and since most of the reactions which are accelerated by the catalytic action of enzymes are either simple[Pg 184] hydrolyses, changes in oxygen content, or other simple decompositions or condensations, which will otherwise proceed so slowly as to be practically negligible, it is customary to speak of the enzyme as "acting upon" the material in question. It should be understood, however, that this is a misstatement, as the enzyme cannot actually initiate a reaction, or "act upon" any substance; it only acts as a catalyzer to accelerate the action of water, oxygen, etc., upon the material in question.
Generally speaking, most enzymes are colloidal in form and, hence, do not diffuse through membranes such as the cell-walls. Some of them perform their characteristic functions only within the cell, or organ, which elaborates them, and can be obtained outside these tissues for purposes of study only by first rupturing the cell-wall or other membrane with which they are surrounded. Such enzymes are known as "intracellular." Others are regularly secreted by glands which discharge them onto other organs, as the stomach or intestines of animals, where they perform their useful functions; or, as in the case of germinating seeds, they move to other parts of the organ, and can be extracted from the tissue by simple treatment with water. These are known as the "extracellular" enzymes.
Enzymes are specific in their action. Any given enzyme affects only a single reaction; or at most acts only upon a single group of compounds which have similar molecular configuration. Usually it is only a single compound whose decomposition is accelerated by the action of a particular enzyme; but there are a few enzymes, such as maltase (which acts on all α-glucosides) and emulsin (which acts on all β-glucosides) which act catalytically upon groups of considerable numbers of similar compounds.
Enzymes, like all other catalysts, act more energetically at increased temperatures; but for each particular enzyme there is an "optimum temperature," (usually between 40° and 65°) above which the destructive effect of the temperature upon the enzyme itself more than offsets the accelerating influence of the increased temperature. At still higher temperatures (usually 80° to 100°) the enzymes are "killed," i.e., rendered permanently inactive. All enzymes are "killed" by boiling the solutions in which they are contained. Dry preparations of enzyme material can withstand somewhat higher temperatures, for somewhat longer periods of time, than can the same enzyme in moist condition or in solution.[Pg 185] When an enzyme has once been inactivated by heating, or "killed," it can never be restored to activity again.
Enzymes are extremely sensitive to acids, bases, or salts, their activity being often enormously enhanced or, in other cases, entirely inhibited, by the presence in the reacting medium of very small amounts of free acids, or bases, or even of certain neutral salts. For example, pepsin, the enzyme of the stomach will act only in the presence of a slightly acid medium and is wholly inactive in a mixture which contains even the slightest amount of free alkaline material; while trypsin, the similar enzyme of the intestine, acts only under alkaline conditions. Practically all enzymes are rendered inactive, but not destroyed, by the presence of either acid or alkali in excess of N/10 strength. Many will act only in the presence of small quantities of certain specific neutral salts; while, on the other hand, other salts are powerful inhibitors of enzyme action. Enzymes often differ from the protoplasm which secretes them in their response to antiseptics, such as toluene, xylene, etc., which inhibit the activity or growth of the cell, but have no effect upon the activity of the enzymes which it contains.
THE CHEMICAL NATURE OF ENZYMES
Nothing is known with certainty concerning the chemical nature of enzymes. Being colloidal in nature, they adsorb carbohydrates, proteins, fats, etc., so that active enzyme preparations often respond to the characteristic tests for these groups of substances; and many investigators have reported what has, at first, seemed to be conclusive evidence that some particular enzyme which they have studied is either a carbohydrate, a protein, or some other type of organic compound. Later investigations have always shown, however, that if the preparation in question be submitted to the digestive action of the enzymes which hydrolyze the particular type of substances to which it is supposed to belong, the material will lose its characteristic protein, or carbohydrate, etc., properties, without losing its specific activity, thus clearly indicating that the substance which responds to the characteristic tests for some well-known type of organic compounds is present as an impurity and is not the enzyme itself.
The present state of knowledge concerning the nature of enzymes seems to indicate that, like the inorganic catalysts, they[Pg 186] may vary widely in chemical composition; and that their tremendous catalytic effects are due, in part at least, to their colloidal nature. This will be better understood and appreciated after the phenomena associated with the colloidal condition have been considered (see the following Chapter).
NOMENCLATURE AND CLASSIFICATION
Since nothing is known of the chemical composition of enzymes, they can only be studied by considering the effects which they produce. This is reflected in the systems which have been adopted for their nomenclature and classification.
As they were first supposed to be proteins, the earlier representatives of the group were given characteristic names ending with the suffix in, similar to that of the proteins. Since this idea has been found to be incorrect, however, a system of nomenclature has been adopted which assigns to each enzyme the name of the material upon which it acts, followed by the suffix ase. Thus, cellulase is the enzyme which accelerates the hydrolysis of cellulose; glucase, that acting upon glucose; amylase, that acting upon starch (amylum), etc.
The substance upon which the enzyme acts (or, strictly speaking, the substance whose hydrolysis, oxidation, or other chemical change, is catalytically affected by the enzyme) is called the substrate.
Most enzymes are catalysts for hydrolysis reactions and are, hence, classed as hydrolytic in their action, and may be spoken of as "hydrolases." Those which accelerate oxidation are called "oxidases"; while those that stimulate reduction reactions are "reductases"; those that aid in the splitting off of ammonia, or amino-acid groups, are "deaminases"; and those that aid in the splitting off of CO2 from COOH groups are "carboxylases," etc.
The hydrolytic enzymes are further subdivided into the sucroclastic (sugar-splitting), or sucrases; the lipoclastic (fat-splitting), or lipases; the esterases (ester-splitting); proteoclastic (protein-splitting), or proteases; etc.[Pg 187]
OCCURRENCE AND PREPARATION FOR STUDY
Enzymes are present in all living matter. In animal tissues, they occur in the largest amounts in those glands or organs where active vital processes take place, as in the brain, the digestive tract, blood, etc. In plants, they may be found in all living cells, and are especially abundant in the seeds, where they serve to render soluble and available to the young plant the stored food materials. The enzymes of moulds, and other parasitic plants, are usually extracellular in type, being secreted for the purpose of making the material of the host plant available to the parasite. Extracellular enzymes are also developed in seeds during germination, in order that the stored food material of the endosperm may be rendered soluble and translocated into the tissues of the growing seedling. But most other plant enzymes are intracellular in type. Hence, in all preparations of plant enzymes for study, or for commercial use, the first step in the process is, necessarily, a thorough rupturing of the cell-walls of the plant material.
The rupturing of the cells may be accomplished in a variety of ways, as follows: (1) mechanical disintegration, as by grinding in a mortar with sharp sand; (2) freezing the material, by treatment with liquid air, then grinding; (3) killing the cells by drying, by treatment with alcohol or acetone, then grinding the mass in a paint mill with toluene; (4) killing the cells by chemicals (sulfuric acid, 0.5 to 1.0 per cent, or other suitable agents) followed by extraction with water; (5) autolysis, or self-digestion, in which the cells are mixed with toluene or some other antiseptic which kills the cells without injuring the enzymes, then the material is minced or ground up and suspended in water containing the antiseptic, until the enzymes dissolve the cell-walls and so escape into the liquid—this process being especially adapted to the preparation of active extracts from yeasts, which contain the necessary cell-wall dissolving enzymes to facilitate autolysis.
Enzymes may be separated out of the aqueous extract obtained from cells ruptured by any of the above methods, by precipitation with alcohol, acetone, or ether, in which they are insoluble; but if this is done, the precipitate must be at once filtered off and rapidly washed and dried, as prolonged contact with these precipitating agents greatly diminishes the activity of most enzymes. Or, they may be adsorbed out of solution on gelatinous, or colloidal, mate[Pg 188]rials, like aluminium hydroxide, or various hydrated clays. If the dry preparations obtained in any of these ways are contaminated by carbohydrates, proteins, etc., these may be removed by treatment with suitable digesting enzymes obtained from the saliva, gastric, and pancreatic juices, and the digested impurities washed out with 60 to 80 per cent alcohol, leaving the enzyme preparation in a purified but still active form.
In any study of the "strength," or possible catalytic effects, of an enzyme preparation, it is necessary, first, to determine what particular reaction it affects, by qualitative tests with various substrate materials, such as starch, sugars, glucosides, proteins, etc., and then to determine quantitatively its accelerating effect upon the reaction in question. The latter may be done by measuring either the time required to carry a unit quantity of the substrate material through any determined stage of chemical change, or the quantity of the substrate which is changed in a unit period of time. It would not be profitable to go into a detailed discussion here of the methods of making these quantitative measurements of enzyme activity. Such discussions must necessarily be left to special treatises on methods of study of enzyme action. It may be said, however, that generally both the qualitative tests for, and the quantitative measurements of, the accelerating influence of enzymes depend upon the observation of some change in the physical properties of the substrate material, such as the optical activity, electrical conductivity, or viscosity, of its solution. In some cases, it is convenient to make an actual quantitative determination of the amount of end-products produced in a given time, as in the inversion of cane sugar, the hydrolysis of maltose, etc., but such determinations necessarily involve the removal of some of the reaction mixture for the purposes of the determinations, and are not, therefore, suitable for the study of the progressive development of the reaction which is being studied.
Enzymes are found in all parts of the animal organism and those which are active in the digestion of food, the metabolism of digested material, the coagulation of blood, etc., have been extensively studied. A discussion of these animal enzymes would be out of place in such a text as this, however, and the following list includes only enzymes which are known to occur in plant tissues. These well-known enzymes will serve as examples of the several general types which have thus far been isolated and studied.[Pg 189]
| Class and Type. | Enzyme. | Substrate. | End-products. | Found in. |
|---|---|---|---|---|
| I. Hydrolases | ||||
| (a) Esterases | Lipase | Fats | Glycerol and fatty acids | Oily seeds |
| (b) Carbohydrases | Sucrase or invertase | Sucrose | Glucose and fructose | Yeasts |
| Maltase | Maltose and all α-glucosides | Glucose, etc. | Barley malt | |
| Dextrinase | Dextrin | Maltose | Malt | |
| Inulase | Inulin | Fructose | Artichokes, etc. | |
| Amylase or diastase | Starch | Maltose | Malt etc. | |
| Cellulase | Cellulose | Maltose | Bacteria | |
| Pectinase | Pectose | Arabinose | Fruits | |
| Cytase | Hemi-celluloses | Mono-saccharides | Nuts, seeds, etc. | |
| (c) Glucosidases | Emulsin | Amygdalin and all β-glucosides | Glucose, etc. | Almond kernels, etc. |
| Maltase | α-glucosides | Glucose, etc. | Barley malt | |
| Myrosin | Sulfur-containing glucosides | Glucose, etc. | Mustard seeds | |
| Rhamnase | Xanthorhamnin | Rhamnose, etc. | Rhamnus spp. | |
| Phytase | Phytin | Inosite and H3PO4 | Bran coats of seeds | |
| (d) Proteases | Erepsin | Proteins | Amino-acids | Many plants |
| Papain | Protein | Amino-acids | Papaws | |
| Bromelin | Protein | Amino-acids | Many plants | |
| Nuclease | Nucleo-proteins | Proteins and nucleic acid | Many plants | |
| [Pg 190] | ||||
| II. Oxidases | ||||
| (a) Catalases | ........ | Hydrogen peroxide | Water and oxygen | Nearly all plants |
| (b) Peroxidases | ........ | Organic peroxides | "Active" oxygen | Nearly all plants |
| (c) Oxidases | ........ | Chromogens Alcohols and phenols |
Pigments Acids |
Many plants Many plants |
| (d) Reductases | ........ | ........ | ........ | Many plants |
| III. Deaminases | Urease | Urea | Ammonia and CO2 | |
| Guanase | Guanine | Xanthine | ||
| Adenase | Adenine | Hypoxanthine | ||
| IV. Carboxylases | ||||
| (a) | ........ | Keto-acids | Aldehydes and CO2 | |
| (b) | ........ | Amino-acids | Amines and CO2 | |
| V. Coagulation enzymes | Pectase | Coagulates pectic bodies | ........ | Fruits |
| VI. Fermentation enzymes | Zymase | Glucose, etc. | Alcohol and CO2 | Yeasts |
| Lactic acid ferment | Fatty acids | Lactic acid | Bacteria | |
| Butyric acid ferment | Fatty acids | Butyric acid | Bacteria | |
The above list includes only the more common and best-known plant enzymes. It seems reasonable to suppose that for every individual type of organic compound which may occur in general plant groups, or even in single species, there is a corresponding enzyme available to affect its physiological alterations. Indeed, new preparations of active enzymes from special types of plants and new evidences of the existence of enzymes in various plant organisms are continuously being reported.
A few of the most common specific representatives of individual groups of enzymes may be briefly described, as follows:
Amylase (or diastase, as it was first named and is still commonly called) is probably the most widely distributed enzyme of plants. It is found in practically all bacteria and fungi; in practically all seeds (it has been found in active form in seeds which were known to be over fifty years old); in all roots and tubers; and in practically all leaves, where it is located in the stroma of the chloroplasts.
It appears to exist in two modifications, known, respectively, as (a) translocation diastase and (b) diastase of secretion. The first form is found in the cells of ungerminated seeds, in leaves, shoots, etc. It remains in the cells where reserve starch is stored and aids in the transformation of starch into soluble materials for translocation from cell to cell. It is active at a lower temperature than the second form, its optimum temperature being 45° to 50°. The second form is secreted by the scutellum, and perhaps by the aleurone cells, of germinating seeds, being produced by special glandular tissue. It aids in the hydrolysis of the starch for the use of the growing embryo. Its optimum temperature is 50° to 55°.
The activity of amylase is accelerated by the presence of small quantities of neutral salts, especially by sodium chloride and disodium phosphate. It acts best in neutral solutions, its activity being inhibited, although the enzyme itself is not destroyed, by the presence of more than minute traces of free mineral acid or alkali.
Sucrase (or invertase) is present in almost all species of yeasts, where it serves to convert unfermentable sucrose into glucose and fructose, which are readily fermentable. Invertase is also present in moulds and other microorganisms; and in the buds, leaves, flowers, and rootlets of those higher order plants which store their carbohydrate reserves in the form of sucrose. It appears that sucrose, while easily soluble, is not readily translocated, or utilized,[Pg 192] by plants until after it has been hydrolyzed into its constituent hexoses.
The optimum temperature for invertase is 50° to 54°; it is killed if heated, in the moist condition, to 70°. Its activity is increased by the presence of small amounts of free acids; but is inhibited by free alkalies.
Zymase is the active alcoholic fermentation enzyme of yeasts. It accelerates the well-known reaction for the conversion of hexose sugars into alcohol and carbon dioxide, namely,
C6H12O6 = 2C2H5OH + 2CO2.
Because of its scientific interest and industrial importance in the fermentation industries, its action has been extensively studied. It acts only in the presence of soluble phosphates and of a coenzyme (see below) which is dialyzable and not destroyed, which is probably an organic ester of phosphoric acid. The significance of the molecular configuration of the hexose sugars in their susceptibility to action by zymase has already been discussed in detail (see page 56).
The optimum temperature for zymase action is 28° to 30°. The enzyme is killed by heating to 45° to 50° in solution, or to 85° if in dry preparation.
Proteases of the erepsin type, i.e., those which break proteins down to amino-acids instead of only to the proteose or peptone stage, as is characteristic of the enzymes of the trypsin type, are widely distributed in plants. Except in the case of the two which occur in large amounts in certain special fruits (papain in papaws, and bromelin in pineapples), they are very difficult to prepare in pure form for study. In general, all proteolytic actions, even when accelerated by active enzymes, proceed much more slowly than do the hydrolyses of carbohydrates or fats. It seems that metabolic changes of the complex protein molecules are much more difficult to bring about and take place much more slowly than do those of the energy-producing types of compounds.
The presence of proteolytic enzymes in most vegetative cells, and in seeds, may be demonstrated, however, by studying the action of extracts of these tissues upon soluble proteins. The best-known example of this type of enzymes is the protease of yeast; but similar ones may be found in germinating seeds. These[Pg 193] vegetable proteases are usually most active in neutral or only faintly alkaline solutions, and their activity is nearly always inhibited by even traces of free acids.
Most laboratory studies of proteolytic enzymes are carried on with preparations of the powerful members of this class of enzymes which are found in the digestive tract of animals, namely, the pepsin of the gastric juice, which acts in the acid medium, in the stomach, and the trypsin of the pancreatic juice, which acts in the alkaline medium of the intestinal tract. But even these powerful proteases require several hours for the transformation of an amount of soluble albumin into its amino-acid constituents which is equivalent to the amount of starch which is hydrolyzed to maltose by diastase in a very few minutes.
Enzymes which govern oxidative changes, known respectively, as catalases and oxidases, are almost universally present in plants. Catalase decomposes peroxides, with the liberation of free oxygen. It is, therefore, necessary to the final step in the process of photosynthesis, as elucidated by Usher and Priestley (see page 26), and serves to prevent the destructive action of hydrogen peroxide upon chlorophyll. The almost universal presence of oxidases in plant tissues has been repeatedly demonstrated. They are present in especially large amounts in tissues which are being acted upon by parasitic fungi or are combating unfavorable conditions of growth. The oxidases, in such cases, seem to be the agents by which the plant is able to stimulate its metabolic activities to overcome the unfavorable environment for its normal development.
In vegetables and fruits, the common browning, or blackening, of the tissues when cut surfaces are exposed to the air has been demonstrated to be due to the catalytic oxidation of the tannins or of certain amino-acids, especially tyrosine, under the influence of the oxidases which are present in the tissues. In fact, most pigmentation phenomena are due to changes in the oxygen content of the chromogens of the cells of the plant, under the influence of the oxidases which are present in the protoplasm of the cells in question. Hence, the oxidases may be said to be the controlling agencies for both the energy-absorbing activities and for respiration in plants.[Pg 194]
THE NATURE OF ENZYME ACTION
The mechanism by which an enzyme accomplishes its catalytic effects has been the object of extensive studies during recent years, especially since the discovery by Büchner that enzymes could be isolated in solutions entirely free from the disturbing influence of growing cells. Several theories concerning the mode of this catalytic action have been advanced. The earliest and simplest of these was that the enzyme simply creates an environment favorable for the particular chemical reaction to take place, as by exposing large surfaces of the substance in question to the action of the hydrolytic, or other effective, agent, by means of surface adsorption of the substrate material on the colloidal enzyme.
However, more recent investigations clearly indicate that there is an actual combination between the substrate material and the enzyme, which combination then breaks down with a resultant change in the substrate material and a freeing of the enzyme for repeated recombination with additional substrate, with the net result that the chemical change in the substrate material is enormously accelerated. That such a combination between substrate and enzyme actually exists has been demonstrated in two different ways: (a) experimentally, by mixing together solutions of an enzyme and of its substrate, each of which is filterable through paper or through a porous clay filter, with the result that the active material in the combined solutions will not pass through these same filters; and (b) mathematically, by a study of the curves representing the reaction velocities of typical reactions which are proceeding under the influence of an enzyme, which show that so long as there is a large excess of substrate material present, the accelerating influence of the catalyst is uniform over given successive periods of time, but that when the quantity of substrate material becomes smaller than that which permits the maximum combining power of the enzyme to be exercised, the reaction velocity immediately slows up.
Again, the fact that the specificity of the action of an enzyme, i.e., the limitation of the action of that enzyme to a specific single compound or group of similar compounds, is definitely related to the molecular configuration of the molecule of the substrate, as has been found to be true in all those cases where the molecular configuration of the substrate material has been established (see[Pg 195] pages 56 to 58), is an added indication that there is some kind of a union between the enzyme and the substrate as a first step in the catalytic process.
As to the nature of this supposed combination of substrate and enzyme, two theories are held. The first is that this union is in the form of an actual molecular combination, or chemical compound, and the other is that it is a purely physical, or colloidal complex. The latter view has by far the greater weight of theoretical and experimental evidence in its support. The relation of electrolytes to the catalytic effect of enzymes, the appearance of the reacting masses under the ultramicroscope, and the effect of heat upon the reacting mixtures, all point to the conclusion that the phenomenon is colloidal rather than molecular in character. This view also makes the remarkable catalytic effects which take place in living protoplasm, which undoubtedly exists in the colloidal condition, much more easily understood. This phase of the matter will be much more apparent after the chapter dealing with the physical chemistry of the protoplasm has been studied.
A further indication that the mechanism of enzyme activity is colloidal in character lies in the fact that, so far as is known, all reactions which are catalyzed by specific enzymes are reversible and the same enzyme will accelerate the velocity of the reaction in either direction, the direction in which the reaction goes being determined by the conditions surrounding the reacting material at the time. It was formerly supposed that enzymes catalyze only decomposition reactions and that the synthetic reactions of living tissues are produced by means of some other force or agency. This view supported the idea of a chemical union of the enzyme with the substrate which, when it breaks down, breaks the molecule of the substrate material into some simpler form, or forms. But it is now known that the reaction which is influenced by the enzyme will be catalyzed in either direction by the specific enzyme which "fits" the particular substrate material at every point of its molecular configuration, as the glove fits the hand. The contrast between this fitting of the enzyme to the entire configuration of the molecule, and the union at a single point or group which is characteristic of chemical linkages, is apparent. As examples of the synthetic action of the same enzyme which, under other conditions, accelerates the decomposition of the same material, there may be cited the demonstrated synthesis of isomaltose from glu[Pg 196]cose by maltase; the production of ethyl butyrate from alcohol and butyric acid; and the synthetic production of artificial fats, by the aid of the pancreatic lipase; and the apparent synthesis of a protein from the same amino-acids which may be obtained from it by hydrolysis under the influence of the same protease, but under different environmental conditions.
ACTIVATORS AND INHIBITORS
The activity of enzymes is strongly influenced by the presence in the solution of other bodies, usually, although not always, electrolytes. This is probably due, in most cases at least, to the action of the electrolyte upon the colloidal condition of the enzyme. All enzymes do not respond alike to the action of the same electrolyte, however. The activity of certain enzymes is enormously increased by the presence of a small amount of acid; while the action of another may be absolutely inhibited by the same acid in the same concentration. Thus, the activity of the amylase found in the endosperm of many seeds is instantly stopped by adding to the solution enough sulfuric acid to make it two-hundredth normal in strength; while the same concentration of acid actually accelerates the activity of some of the proteases.
Formaldehyde, hydrocyanic acid, and soluble fluorides usually inhibit both the activity of a cell and of the enzymes which it contains; while other antiseptics, such as toluene, xylene, etc., prevent the growth of the cell, or organism, without interfering with the activity of the enzymes which may be present. By the use of this latter type of antiseptics, it is possible to distinguish between chemical changes which are involved in the actual development of a cell and those which can be brought about in other media by means of the enzymes which are contained in the cell.
Any substance which increases the catalytic activity of an enzyme is known as an "accelerator," or "activator"; while one which prevents this activity is called an "inhibitor," or "paralyzer."
A type of accelerating influence quite different from that of electrolytes is found in the effect of certain amino-acids upon enzyme action. The influence of small amounts of asparagine in enormously increasing the hydrolytic effect of amylase is an exam[Pg 197]ple. There is no known explanation for this type of activation of the enzyme.
The influence of activators, or inhibitors, in providing favorable or unfavorable conditions for the action of an enzyme, should not be confused with the relation to the enzyme itself of what are known as "coenzymes" and "antienzymes," discussed in the following paragraph.
COENZYMES AND ANTIENZYMES
In the cases of many enzymes of animal tissues, it has been found that they are absolutely inactive unless accompanied by some other substance which is normally present in the gland, or protoplasm, which secretes them. Thus, the bile salts are absolutely necessary to the activity of trypsin, in its characteristic protein-splitting action. Such substances are known as "coenzymes." They can usually be separated from their corresponding enzymes by dialysis, the coenzyme passing through the parchment membrane. Such coenzymes are not killed by boiling the dialyzate, and the activity of the enzyme is restored by adding the boiled dialyzate to the liquid which remains within the dialyzer.
The best known example of a coenzyme in plant tissues is in connection with the activity of the zymase of yeast cells. If yeast juice be filtered through a gelatin filter, the colloidal enzymes which are left behind are entirely inactive in producing fermentation, but may be restored to activity again by mixing with the filtrate. An examination of this filtrate, which contains the coenzyme for zymase, shows that it contains soluble phosphates and some other substance whose exact nature has not yet been determined, both of which are necessary to the activity of the zymase. The phosphates seem to enter into some definite chemical combination with the substrate sugars, while the other coenzyme seems to be necessary in order to make possible the final breaking down of the sugar-phosphate complex by the zymase. This phenomenon of coenzyme relationship is not very frequently observed in plant enzyme studies, probably because the coenzyme (if there be such, in the case which is under observation) usually accompanies the enzyme itself through the various processes of extraction and purification of the material for study. However, care must be[Pg 198] taken in all cases when dialysis is employed, to see that a possible coenzyme is not separated from an otherwise active preparation.
An entirely different type of phenomenon is that exhibited by "antienzymes." These are found in the various intestinal worms which live in the digestive tracts of animals; and prevent the digestive action of the enzymes of the stomach and intestines upon these worms. Probably similar "antienzymes" are located in the mucous linings of the intestinal tract itself, and serve to prevent the auto-digestion of these organs by the active enzymes with which they are almost continually in contact.
The difference between an antienzyme, which protects material which would otherwise be subject to the attack of an enzyme, and an inhibitor, which renders the enzyme itself inactive, is apparent.
So far as is known, however, no such substances as antienzymes are present in plant tissues; although the question as to why the proteoclastic enzymes which are elaborated by a given mass of protoplasm do not attack the protoplasm itself, might well be raised.
ZYMOGENS
It is apparent that, since enzymes are produced by protoplasm for the special needs of any given moment or stage of development, there must be a preliminary stage, or condition, in which they do not exert their characteristic catalytic effect. When in this stage, the compound is known as "proenzyme," or "zymogen." In this stage, it is inactive, but can be made to exhibit its catalytic effect, usually by bringing it into contact with a suitable activator. When once so activated, however, it cannot be returned again to the inactive state.
This phenomenon has been studied in connection with the zymogens of the digestive proteases, pepsin and trypsin. Trypsinogen may be rendered active by contact with either calcium salts or with another substance (apparently itself an enzyme) known as enterokinase, which is secreted in the intestinal tract.
Similarly, proenzymes have been reported as occurring in numerous plant tissues. These proenzymes are believed to be present in the plant cells in the form of definite characteristic granules, which may be observed under the microscope, and which disappear when the enzyme becomes active. Thus, "proinu[Pg 199]lase" has been reported as occurring in artichoke tubers: "prolipase," in castor beans; "proinvertase," in several species of fungi; and, probably, "prooxidase," in tobacco leaves. In the case of the last-named zymogen, it has been observed that after the zymogen has been once activated, as in response to the need for increased activity due to the entrance of the germs of certain leaf-diseases, it can once again produce a second supply of the enzyme, but the process cannot again be repeated.
Calcium salts, or very dilute acids, are usually energetic activators of proenzymes.
PHYSIOLOGICAL USES OF ENZYMES
There can be no doubt that enzymes exert a tremendously important influence in vital phenomena, by determining the rate at which the chemical changes which are involved in these phenomena shall proceed. Since they do not initiate reactions, and since they may catalyze reversible reactions in either direction, it cannot be said that they determine the type of reactions which will take place in any given mass of protoplasm; but, undoubtedly, they do exert a determining influence upon the rate at which the reaction will proceed, after the protoplasmic activity has determined the direction in which it shall go.
Without the intervention of these catalyzing agents, it would be impossible for reactions between these non-ionized organic components of the cell contents to come to completion with anything like the marvelous rapidity with which these changes must take place in order to permit the organism to grow, to perform its necessary vital functions, or to adjust itself to the changes in its environmental conditions.
Since the number of different reactions which take place within a living cell is very great, and since these chemical changes are extremely variable in type, it follows that the number of different enzymes which must exist in either a plant or an animal organism is likewise very large. For example, fourteen different enzymes have been isolated from the digestive system, and at least sixteen from the liver, of animals. They are universally present in living protoplasm of every kind, from the most minute bacterium to the largest forest trees, in the plant kingdom; and from the amœba to the whale, in animals.[Pg 200]
While there is a great variety of enzymes which may be produced by a single individual organism, the same enzyme may be found in the greatest variety of organisms; as, for example, the protease trypsin, which has been found in several species of bacteria, in the carnivorous plant known as "Venus' Fly Trap," and in the human pancreas, as well as that of all other animals.
FURTHER STUDIES NEEDED
From the discussions which have been presented in this chapter, it is apparent that the enzymes play a tremendously important part in vital phenomena, by controlling the rate at which the biochemical reactions take place in the cells of the living organism.
The means by which the protoplasm elaborates these all-important chemical compounds are as yet absolutely unknown. Even the nature of the enzymes themselves is still a matter of speculation and study. Much intensive study is needed and should be given to these matters, for the purpose of elucidating the methods by which the enzymes accomplish their remarkable catalytic effects, and, if possible, the actual chemical nature of the enzymes themselves. It is conceivable, of course, that if the latter object of these studies should ever be reached, it might be possible to synthetize enzymes artificially, and so to develop a means for the artificial duplication of the synthesis of organic compounds with the same velocity that this is done in the plant cells. Such a result would have a scientific interest fully as great as did Wöhler's artificial synthesis of urea, which proved that there is no essential difference in character between the compounds which are the products of living organisms and those which are produced in the laboratory; and, at the same time, might have an immensely more important practical bearing, since it would lead the way to the artificial production of the carbohydrates, proteins, fats, etc., for which we are now dependent upon plant growth as the source of these materials for use as human food.
References
Bayliss, W. M.—"The Nature of Enzyme Action," 186 pages, Monographs on Biochemistry, London, 1919 (4th ed.).
Euler, H., trans. by Pope, T. H.—"General Chemistry of the Enzymes," 319 pages, 7 figs., New York, 1912.[Pg 201]
Effront, J., trans. by Prescott, S. C.—"Enzymes and their Application,—Enzymes of the Carbohydrates," 335 pages, New York, 1902.
Effront, J., trans. by Prescott, S. C.—"Biochemical Catalysts in Life and Industry—Proteolytic Enzymes," 763 pages, New York, 1917.
Green, J. R.—"The Soluble Ferments and Fermentation," 512 pages, Cambridge, 1901, (2d ed.).
Grus, J.—"Biologie und Kapillaranalyse der Enzyme," 227 pages, 58 figs., 3 plates, Berlin, 1912.
Harden, A.—"Alcoholic Fermentation," 156 pages, 8 figs., Monographs on Biochemistry, London, 1914.
Plimmer, R. H. A.—"The Chemical Changes and Products Resulting from Fermentations," 184 pages, London, 1903.
Oppenheimer, C., trans. by Mitchell, C. A.—"Ferments and their Actions," 343 pages, London, 1901.[Pg 202]
CHAPTER XV
THE COLLOIDAL CONDITION
Reference has frequently been made, in preceding chapters, to the fact that proteins, enzymes, lipoids, etc., exist in the protoplasm of plants and animals in the colloidal condition. The properties and uses of these compounds by plants depend so much upon this fact that, before proceeding to the consideration of the actual physical chemistry of protoplasm itself, it will be appropriate and profitable to give some attention to the nature and significance of the colloidal condition of matter and of some of the phenomena which grow out of it.
Every discussion of the colloidal condition in general properly begins with reference to the work of the English physicist, Thomas Graham, who carried on his investigations of the so-called "colloids" through a period of forty years, beginning with 1851. His most important results were published, however, from 1861 to 1864. Graham studied the diffusibility of substances in solution through the parchment membrane of a simple dialyzer. As a result of his earlier investigations, he divided all the chemical compounds which were known to him into two groups, which he called "crystalloids" and "colloids," respectively, the first including those substances which readily diffused through the parchment membrane and the second those which diffused only very slowly or not at all. He at first thought that crystalloids are always inorganic compounds, while colloids are of organic origin. He soon learned, however, that this distinction in behavior is not always related to the organic or inorganic nature of the compound. He further discovered that the same individual chemical element or compound may exist sometimes in crystalloidal, and sometimes in colloidal, form. This latter discovery led to the conclusion that diffusibility depends upon the condition, rather than upon the nature, of the material under observation.
As a result of the long series of investigations which were stimulated by Graham's work, the modern conception is that dif[Pg 203]fusibility is a condition of matter when in minute subdivision, or in solution, in some liquid, as contrasted with its state, or condition, when existing alone. That is, the state of a substance may be either gaseous, liquid, or solid; and its condition when in solution may be either crystalloidal or colloidal. Substances which are in crystalloidal form, in true solution, exist there in molecular or ionized condition; but, as will be pointed out below, when in the colloidal condition they exist in aggregates which are somewhat larger than molecules, but not large enough to be visible as individual particles under the ordinary microscope, even under the highest magnification which has yet been obtained. Colloidal particles are, however, generally visible under the Zigmondy "ultramicroscope." (See below.)
The use of the word "colloid" as a noun, or as the name for a substance which is in the colloidal condition, is of the same nature as the use of the words "gas," "liquid," and "solid," in such statements as "ice is a solid," "water is a liquid," or "steam is a gas," etc.; i.e., the noun represents a state or condition rather than an actual object or thing. Hence, the expression "enzymes are colloids," means only that enzymes exist in the colloidal condition, and not that enzymes represent a definite type of substances having the group name "colloids."
THE COLLOIDAL CONDITION A DISPERSION PHENOMENON
When one substance is distributed through the mass of another substance, the mixture is said to be a "two-phase system," composed of the dispersed phase, or substance, and the dispersion medium, or continuous phase, through which the other substance is distributed. The following examples illustrate the possibilities of such two-phase systems:
- Dispersion medium a gas.
- Disperse phase a liquid—mist in the air.
- Disperse phase a solid—smoke or dust in air.
- Dispersion medium a liquid.
- Disperse phase a gas—foams.
- Disperse phase a liquid—emulsions.
- Disperse phase a solid—suspensions.
- Dispersion medium a solid.
- Disperse phase a gas—solid foams, pumice stone, etc.
- Disperse phase a liquid—liquid inclusions in minerals.
- Disperse phase a solid—alloys, colored glass, etc.
Although the same general principles of physical chemistry apply to all two-phase systems, the term "colloidal condition" is commonly used only in connection with a particular type of dispersions, in which the dispersion medium is a liquid and the dispersed material is either a solid or a liquid.
Thorough and careful studies have shown that when a solid or a liquid is introduced into another liquid, and becomes dispersed or distributed through it, the mixture may be either a true solution, a colloidal solution, or a mechanical suspension. The characteristic differences between these three conditions may be tabulated as follows: although the significance of some of the phrases used will not be apparent until the phenomena in question have been considered in some detail.
| True Solutions. | Colloidal Solutions. | Suspensions. |
|---|---|---|
| (a) Particles of the disperse phase are: | ||
| In molecular subdivision | In colloidal subdivision | In mechanical subdivision |
| Invisible | Visible under "ultrascope" | Visible under microscope or to naked eye |
| Less than 1µµ in diameter[6] | 1µµ to 1µ in diameter | Greater than 1µ in diameter |
| In molecular motion | In Brownian movement | Do not pass through filters or parchment |
| Pass through filters and parchment membranes | Pass through filters but not through parchment | In gravitational movement |
| (b) The system exhibits: | ||
| High osmotic pressur | Low osmotic pressure | No osmotic pressure |
| Transparency | "Tyndall phenomenon" | Is generally opaque |
| No gel-formation | Forms gels | No gel-formation |
[6] 1µ is one-thousandth of a millimeter; 1µµ is one-thousandth of a µ, or one millionth of a millimeter.
It is recognized by all students of these matters that it is not possible to draw a sharp dividing line between these three types of conditions, and that they shade into each other, in many cases; but in general it may be said that a colloidal solution is one in which the dispersed particles are usually between 5µµ and 200µµ in diameter, are difficultly or not at all diffusible through the mem[Pg 205]brane of a simple dialyzer, cannot be filtered out of solution, do not settle out under the action of gravitation, and are visible only under the "ultramicroscope"; and one which has certain peculiar optical, osmotic, and other physical and chemical properties. Since colloidal particles are very minute in size, they possess very large relative surface areas as compared with their total mass or volume, very high surface tension, and a relatively high surface energy as compared with their total, or molecular, energy. These properties bring into play, in a substance which is in the colloidal condition, in a remarkable degree, all the phenomena which are associated with surface boundaries between solids and liquids, liquids and gases, etc.
The properties arising out of the colloidal condition are of such tremendous importance in connection with the vital phenomena exhibited by cell protoplasm that it is necessary to give some detailed consideration to them here. Many large volumes dealing with this condition of matter have been written, and it is very difficult to condense even the most important facts concerning it into a few pages, but an attempt has been made to present in this brief summary the most essential facts and principles involved in the colloidal phenomena.
NOMENCLATURE AND CLASSIFICATION
Colloidal mixtures may exist in two different forms: one, in which the mixture is fluid and mobile, like a true solution, is known as a "sol"; and the other, which is a semi-solid, or jelly-like, form, is known as a "gel." Sols may be easily converted (or "set") into gels, by changes of temperature or of the electrolyte content, or by changes in the concentration of the mixture, etc., and in most cases gels can be converted again into sols. In some cases, however, gel-formation is irreversible, the gels are permanent and cannot be changed back again into sols by any known change in environmental conditions.
Depending upon whether the liquid dispersion medium is water, alcohol, ether, etc., sols are known as "hydrosols," "alcosols," "ethersols," etc.; and gels as "hydrogels," "alcogels," etc.
Sols in which the disperse phase is a solid are known as "suspensoids"; while those in which it is a liquid are "emulsoids." Thus, sols of most inorganic compounds, of dextrin, gelatin, and[Pg 206] (probably) of casein, etc., are suspensoids; while sols of egg-albumin, of oils, etc., are emulsoids. The classification of these substances into suspensoids and emulsoids is, however, more a matter of convenience than of real difference in composition, since it is practically impossible to say whether many of the organic substances which normally exist in colloidal form are themselves liquids or solids, when in the non-dispersed form.
CONDITIONS NECESSARY TO THE FORMATION OF SOLS
Suspensoids differ from mechanical suspension of solids in a liquid in that in the latter the solid particles settle toward the bottom of the mixture, because of the effect of the attraction of gravity upon them. The rate at which such particles settle depends upon the size and density of the particle and the viscosity of the liquid, and can be roughly calculated from the formula for Stokes' law for the rate of falling of a spherical body in a liquid. This formula is
- 2r2(s - s´)g
- V = —————
- 9n
- V = velocity of the falling body, in millimeters per second;
- r = radius of the particle, in millimeters;
- s = specific gravity of the solid;
- s´ = specific gravity of the liquid;
- g = the attraction of gravity, in dynes;
- n = the viscosity of the liquid.
For example, if this formula be applied to determine the rate at which the particles of gold of the size of those in a red gold sol would settle, if they were in mechanical suspension in water (r = 10µµ, or one-ten-thousandth of a millimeter; s = 19.3; s´ = 1; g = 980, and n = 0.01), it will be found that such particles will settle at the rate of approximately 0.0146 millimeter per hour, or a little over 10 mm. (0.4 inch) per month. Hence, the settling of such particles, if in mechanical suspension, would be measurable, although very slow. Shaking up the suspension would cause the particles to rise through the liquid again. But in a gold sol, or suspensoid, which contains particles of gold of the size used in this calculation, the gold particles do not settle, even at the slow rate as calculated above. They remain uniformly distributed through[Pg 207]out the liquid for an indefinite period or time. The reason for this phenomenon undoubtedly lies in the fact that these minute particles carry an electric charge, which, is of the same sign for all of the particles and results in a repellent action which keeps the particles in constant motion. This constant motion may easily be conceived to keep the particles uniformly distributed throughout the liquid, just as constant shaking would keep those of a mechanical suspension uniformly distributed through the mixture.
The sign of the electric charge on the particles of a sol may be either negative or positive, depending upon the chemical nature and dielectric constants of the two phases of the system. The proportion of the total electric charge of the system which is of the opposite sign to that borne by the dispersed particles is, of course, borne by the liquid which constitutes the other phase. The origin of this electric charge on the colloidal particles is, as yet, not known with certainty; but it seems probable that it is due to a partial ionization of these small particles, similar to, but not so complete as, that which takes place when compounds which are soluble go into true solution in water, or other solvents which bring about the dissociation of dissolved substances.
The conditions necessary to bring a solid substance into a colloidal mixture with some liquid, or, in other words, to produce a suspensoid sol, require that the proportion of liquid to solid shall be large and some means of disintegrating the material which is to be dispersed into very fine particles. Many common chemical reactions, if carried out in very dilute solutions, result in the production of sols, especially if a small amount of some emulsoid is present in the reacting mixture; sols produced in this way are very stable, and the emulsoid which is used in stabilizing the sol is known as a "protective colloid." Direct methods of disintegration; such as reduction by chemical agents, discharge of a strong electrical current through the substance which is to be dispersed while it is submerged in the liquid, alternate treatment of finely ground material with alkali and acid so as to frequently change the electric charge, etc., are utilized for bringing inorganic compounds into the colloidal state.
Suspensoids usually contain less than 1 per cent of the solid dispersed through the liquid. In fact, extreme dilution is one of the necessary conditions for suspensoid-formation.
Emulsoids are much more easily produced than are suspensoids.[Pg 208] The property of forming an emulsoid seems to be much more definitely a characteristic of the substance in question than does the formation of sols from solids which, under other conditions, may form true solutions. This difference may be due to the fact that the liquids which easily form emulsoids (usually those of organic origin) have very large molecules, so that the transfer from molecular to colloidal condition involves much less change in such cases than it does in the case of solid (inorganic) substances of relatively low molecular weight. This view of the matter is further borne out by the fact that solids which have very large molecules (generally of organic origin) take on the colloidal form much more readily than do those of small molecular size.
At the same time, a given liquid may form a true emulsoid when introduced into one other liquid and a true solution when introduced into another. Thus, soaps form emulsoids with water (true hydrosols); but dissolve in alcohol to true solutions, in which they affect the osmotic pressure, the boiling point of the liquid, etc., in exactly the same way that the dissolving of other crystalloids in water affects the properties of true aqueous solutions. Again, ordinary "tannin," when dissolved in water, produces a sol, which froths easily, is non-diffusible, etc.; but when dissolved in glacial acetic acid, it produces a true solution.
The concentration of the disperse phase may be much greater in the case of emulsoids than it can be in suspensoids. This is probably because the dispersed particles do not carry so large an electric charge and are not in such violent motion.
GEL-FORMATION
The one property which most sharply distinguishes sols from true solutions is their ability to "set" into a jelly-like, or gelatinous semi-solid, mass, known as a "gel," without any change in chemical composition, or proportions, of the two components of the system. In the gel, the two components are still present in the same proportions as in the original sol; but the mixture becomes semi-solid instead of fluid in character. Thus, an agar-agar sol containing 98 per cent of water sets into a stiff gel; while many other gels which contain 90 to 95 per cent of water can be cut into chunks with a knife and no water will ooze from them. The water is not in chemical union with the solid matter in the form of[Pg 209] definite chemical hydration, however, as the same gel is formed with all possible variations in the water content.
Gels may be either rigid, as in the case of those of silicic acid, etc., or elastic, as are those of gelatin, egg-albumin, agar-agar, etc. The latter are the common type of gels among organic colloids. They can be easily changed in shape, or form, without any change in total volume.
In gel-formation, the two phases of the system take a different relationship to each other. The disperse, or solid, phase becomes associated into a membrane-like, or film, structure, surrounding the liquid phase in a cell-like arrangement. That is, the whole mass takes on a structure similar to a honeycomb except that the cells are roughly dodecahedral in shape, instead of the hexagonal cylinders in which the bees arrange their comb cells, in which the original disperse phase constitutes the cell-walls and the original liquid, or continuous phase, represents the cell-contents. The cells of an elastic gel resemble closely the cells of a plant tissue in many of their physical properties. They are roughly twelve-sided in shape, as this is the form into which elastic spherical bodies are shaped when they are compressed into the least possible space.
Imbibition and Swelling of Gels.—When substances which are natural gels, such as gelatin, agar-agar, various gums, etc., are submerged in water, they imbibe considerable quantities of the liquid and the cells become distended so that the mass of the material swells up very considerably. This swelling will take place even against enormous pressures. For example, it has been found that the dry gel from sea-weeds will swell to 330 per cent of its dry volume, if immersed in water under ordinary atmospheric pressure; but that it will increase by 16 per cent of its own volume when moistened, if under a pressure of 42 atmospheres.
During the swelling of gels by imbibition of water, the total volume of the system (i.e., that of the original dry gel plus that of the water absorbed) becomes less. For example, a mixture of gelatin and water will, after the gelatin has swelled to its utmost limit, occupy 2 per cent less space than the total volume of the original gelatin and water. It has been computed that a pressure equivalent to that of 400 atmospheres would be necessary to compress the water to an extent representing this shrinkage in volume.[Pg 210]
On the other hand, gels when exposed to the air lose water by evaporation, shrink in volume, and finally become hard inelastic solids, as in the case of the familiar forms of glue, gelatin, agar-agar, gum arabic, etc.
The difference in the relation of gels and that of non-colloidal solids to water may be illustrated by the different action of peas, beans, etc., and of a common brick, when immersed in water. Each of these substances, under these conditions, absorbs, or "imbibes," water; but the peas and beans swell to more than twice their original size and become soft and elastic, while the brick undergoes no change in size, elasticity, or ductility. In all cases of colloidal swelling, the swollen body possesses much less cohesion, and greater ductility, than it had before swelling. The essential difference in the two types of imbibition is that in the case of the non-swelling substances the cohesion, or internal attraction of the molecules of the material, is too great to permit them to be forced apart by the water; while in colloidal swelling, the particles are forced apart to such an extent as to make the tissue soft and elastic. It is possible, of course, to make this separation go still further, until there is an actual segregation of the molecules, when a true solution is produced; for example, gum arabic when first treated with water swells into a stiff gel, then into a soft gel, and finally completely dissolves into a true solution.
Reversibility of Gel-formation.—In some cases, the change of a sol to a gel is an easily reversible one. Glue, gelatin, various fruit jellies, etc., "melt" to a fluid sol at slightly increased temperatures and "set" again to a gel on cooling, and the change can be repeated an indefinite number of times. On the other hand, many gels cannot be reconverted into sols; that is, the "gelation" process is irreversible. For example, egg-albumin which has been coagulated by heat cannot be reconverted into a sol; casein of milk when once "clotted" by acid cannot again be converted into its former condition, etc. Irreversible gelation is usually spoken of as "coagulation." Some coagulated gels, by proper treatment with various electrolytes, etc., can be converted into sols, the process being known as "peptization"; but in such "peptized" hydrosols, the material usually exists in a different form than originally, having undergone some chemical change during the peptization, and the coag[Pg 211]ulation and peptization cannot be repeated, that is, the process is not a definitely reversible one.
Importance of Gel-formation.—From the physiological point of view, gel-formation is undoubtedly the most important aspect of colloidal phenomena. In the first place, the ability to absorb and hold as much as 80 to 90 per cent of water in a semi-solid structure is of immense physiological importance. In no other condition can so large a proportion of water, with its consequent effect upon chemical reactivity, be held in a structural, or semi-solid, mass. But a vastly more significant feature of the conditions supplied by the gel lies in the fact that the non-water phase, or phases, of the system are spread out in a thin film, or membrane, thus giving it enormous surface as compared with its total volume. This effect is easily apparent if one thinks of the enormous surface which is exposed when a tiny portion of colloidal soap is blown out into a "soap-bubble" several inches in diameter. This condition brings into play all the phenomena resulting from surface boundaries between solids and liquids, liquids and liquids, liquids and gases, etc., from surface tension, surface energy, etc. Among these effects may be cited those of adsorption, increased chemical reactivity due to enlarged areas of contact, permeability and diffusion, etc., the importance of which in the vital phenomena of cell-protoplasm will be discussed in detail in the following chapter.
GENERAL PROPERTIES OF COLLOIDAL SOLUTIONS
Non-diffusibility.—The most characteristic property of all sols is the failure of the suspended particles to pass through a parchment, or any similar dialyzing membrane.
Visibility under the "Ultramicroscope."—The particles of a sol, in contrast with the molecules of a true solution, are visible as bright scintillating points under the ultramicroscope. This is a modification of the type of dark-field illumination of the ordinary microscope, as applied to microscopic studies, in which the solution to be studied is contained in a small tube or box of clear glass which is mounted on the stage of an ordinary microscope and instead of being illuminated from below by transmitted light is illuminated by focusing upon it the image of the sun, or of some other brilliant source of light such as an electric arc,[Pg 212] by passing the rays from the source of light through a series of condensing lenses which are adjusted at the proper distance and angles to bring the image of the illuminating body within the tube containing the substance which is to be examined and in the line of vision of the microscope. Obviously, this results in intense illumination of any particles in the solution which come within this brilliant image of the sun, or arc, and therefore renders visible particles which are of less diameter than the wave-length of ordinary light (450µµ to 760µµ for the visible spectrum) and, hence, are not visible by the ordinary means of illumination in the direct line of vision. It will be apparent that what is seen in the field of the ultramicroscope is not the particles themselves, but rather the image of the sun (or other illuminating body) falling upon the particles which come within the image, just as one does not see the paper but only the image of the sun when the rays from the sun are brought to a focus upon a sheet of paper through any ordinary convex lens, or "burning glass." Hence, the ultramicroscope gives no idea of the shape, color, or size of the particles upon which the image falls; but it does permit the counting of the number of particles within a given area, and a study of their movements, from which it is possible, by mathematical computations, to calculate the relative size of the particles themselves. Repeated studies have shown that particles of the sizes between 5µµ and 250µµ in diameter, which are visible under the ultramicroscope, are sufficiently small to bring about the surface phenomena which are known as properties of colloidal solutions. Further, the ultramicroscope permits the observation of the growth, or disintegration, under various chemical reagents, of the individual colloidal particles, which appear as scintillating points in the field of the microscope; and the study of changes in relationships during gel-formation, peptization, etc.
The "Tyndall Phenomenon."—Colloidal solutions exhibit this phenomenon; that is, if a bright beam of light be passed through a sol which is contained in a clear glass vessel having parallel vertical sides, and the solution be viewed from the side, it appears turbid and often has a more or less bluish sheen. This effect is due to the small particles in the sol, of polarizing the light which is reflected from them, the blue rays being bent more than are those in the other part of the spectrum. The Tyndall phenomenon is similar in its effect in making the tiny particles of the sol visible[Pg 213] to the illumination of the dust particles in the air of a darkened room when a ray or narrow beam of light passes through it. In a true molecular solution, the particles are too small to be visible by this mode of illumination.
Other Optical Properties.—Sols are generally translucent and opalescent; many of them are highly colored, some of the sols of gold, platinum and other heavy metals possessing particularly brilliant colors. In general, metallic suspensoids are red, violet, or some other brilliant color; while inorganic suspensoids are bluish white, and emulsoids generally blue to bluish white.
Formation of Froth, or Foam.—Colloidal solutions, especially those of the natural proteins, fats, glucosides, gums, and the artificial soaps, have a strong tendency to produce froth, or foam, when shaken; this being due to the enormous surface tension resulting from the finely divided condition of the dispersed material.
Low Osmotic Pressure.—All colloidal solutions exhibit a very low osmotic pressure; the freezing point of the dispersion medium is lowered only very slightly and its boiling point is only very slightly raised by the presence of the dispersed particles in it.
Precipitation by Electrolytes.—Sols of all kinds are precipitated, or caused to form gels, by the addition of electrolytes, since these cause a disturbance of the electric charge on the dispersed particles, to which the colloidal condition is due. In the case of most emulsoids and of a few of the suspensoids, this change converts the mass into a stiff gel; but in that of many of the metallic suspensoids, the dispersed particles are gathered together into larger aggregates, which settle out of the liquid in the form of a gelatinous precipitate. In the latter case, the effect is usually spoken of as "precipitation" by electrolytes; while in the former, it is called "coagulation," or "gelation."
The effectiveness of the various electrolytes in bringing about this change is proportional to their valency; bivalent ions are from 70 to 80 times, and trivalent ions about 600 times as effective as monovalent ions.
Further, all sols in which the dispersed particles carry a charge of the opposite sign likewise precipitate both suspensoids and emulsoids.
A demonstration of the presence of an electric charge on the particles of a sol and a determination of its sign can be made by[Pg 214] placing the solution in a U tube, with a layer of distilled water above the sol in each arm of the tube, and then passing an electric current through the contents of the tube, keeping the electrodes in the distilled water, so that the migration of the particles toward one pole or the other can be observed by their appearance in the clear water at that end of the tube; or by passing an electric current through the observation chamber of an ultramicroscope, in which the solution under examination has been placed, and observing the migration of the particles across the field toward either one or the other (positive or negative) electrode.
Emulsoids and suspensoids differ in their properties in the following respects. Suspensoids are always very dilute, containing less than 1 per cent of the dispersed solid; while emulsoids may be prepared with widely varying proportions of the two component liquids. Suspensoids have a viscosity which is only slightly greater than that of the liquid phase when it exists alone, and their viscosity varies with the proportion of dispersed solid which is present in the sol; while emulsoids have a very high viscosity in all cases. Emulsoids usually form stiff gels when treated with electrolytes; while suspensoids more commonly yield gelatinous precipitates under the same conditions.
Suspensoids and emulsoids which carry electric charges of opposite sign mutually precipitate each other. But emulsoids often protect suspensoids from precipitation by electrolytes, by forming a protective film around the particles of the suspensoids, which prevents the aggregation of the particles into the precipitate form.
ADSORPTION
If a sol be precipitated or coagulated by the action of an electrolyte, substances which may be present in solution in the liquid of the sol are carried out of solution and appear in the gel or precipitate. This phenomenon is known as "adsorption," which means the accumulation of one substance or body upon the surface of another body, as contrasted with "absorption," which means the accumulation of one substance within the interior of another. Since substances which are in the colloidal form have very large relative surface areas, it follows that the opportunity for surface adsorption on colloidal materials is very great.[Pg 215]
Surface adsorption is a common phenomenon. It was extensively studied by the physicist, Willard Gibbs, who showed that adsorption will take place whenever the surface tension of the adsorbing body will be lowered by the concentration in its surface layer of the material which is available in the solution or other surrounding medium.
As applied to colloidal phenomena, adsorption may be exhibited in either one of four different ways, as follows: (1) A crystalloidal substance which is in solution may be adsorbed on the colloidal particles of a hydrosol, so that if the mixture be dialyzed, or filtered through a so-called "ultrafilter" (i.e., a filter with pores so small that it will retain colloidal particles) the dissolved crystalloid will remain with the separated colloidal particles, or the dissolved crystalloid will not react chemically as it would in a free solution. For example, if to a solution of methylene blue, which dyes wool readily, there be added a small quantity of albumin (a colloidal substance), the dye is adsorbed by the albumin and will no longer color wool with anything like the same readiness. (2) During gel-formation, electrolytes and other soluble substances which may be present in solution in the liquid may adsorbed out of the solution and appear in the gel. For example, a precipitate of aluminium hydroxide, or of silicic acid, is nearly always contaminated with the soluble salts which are present in the solution, and can be prepared in pure form only by repeated filtering, redissolving, and reprecipitating. (3) Colloidal substances may be removed from sols by being adsorbed upon porous materials like charcoal, fuller's earth, hydrated silicates, etc. For example, animal charcoal (or bone black) is used commercially for the clarification of sugar solutions, because it adsorbs out of these solutions the colloidal proteins, coloring matters, etc., with which they are contaminated. (4) Finally, colloids mutually adsorb each other, as in the case of the "protective colloids" previously referred to.
Certain characteristics of adsorption phenomena are of interest and importance from both the physiological and the industrial point of view. The following may be mentioned: (a) Amount of adsorption. Relatively more material is adsorbed out of dilute solutions than out of more concentrated ones. An increase of ten times in the concentration of the dissolved material results in only four times as much adsorption by the colloidal substance which[Pg 216] may be introduced into the two solutions. In this, adsorption differs from chemical action, as the latter is proportional to the concentration of the reacting material which is present in the solution. (b) Adsorption out of different liquids, by the same adsorbing body, is different in amount. It is usually greatest out of water. Hence, many dyes may be adsorbed out of water by charcoal, porous clay, etc., and if the latter be then introduced into alcohol, or ether, the dye goes back into solution in these latter liquids. This process is often used industrially and in the laboratory for the purification of such substances when they are present in impure form in aqueous solutions. (c) Selective adsorption. Different substances are not adsorbed out of the same solvent to the same extent by the same adsorbing agent. Advantage is taken of this fact when filter paper is used in the so-called "capillary analysis" to separate different dyes, or other colloidal materials which have been stained different colors, into alternate layer by reason of the different rate at which the paper adsorbs the different materials out of the solution in which they are present together. (d) Similar relative adsorption by different adsorbing agents. Although different adsorbing agents may possess varying active surfaces and hence, variable adsorbing power, or rates of adsorption, they adsorb the same relative amounts of different materials; i.e., if substance A adsorbs more of X than it does of Z out of any given solution, substance B will likewise adsorb more of X than of Z out of the same solution; although the actual amounts adsorbed by A may be quite different from those adsorbed by B.
CATALYSIS AFFECTED BY THE COLLOIDAL CONDITION
The velocity of a chemical reaction is the net result of opposing influences. It is directly proportional to the chemical affinity of the reacting bodies and inversely proportional to the so-called "chemical resistance." The first factor, chemical affinity, is not easily measured, as it depends upon both the mass of the reacting molecules, atoms, or ions, and their attraction for each other. But if, as the result of chemical affinity, a reaction takes place, it is evident that the time required for its completion (which measures the velocity of the reaction) is made up of two separate periods. The first is the time required for the reacting molecules to come into contact; and the second is that required for the molecular rear[Pg 217]rangement which constitutes the reaction. Clearly, the time required for the substances to come into molecular contact will be greatly diminished if they are mutually adsorbed in large quantities on the extended surface area of some colloidal catalyst which is present in the mixture rather than scattered throughout its entire volume. The application of this principle to the catalysis of hydrolytic reactions is not apparent, if it is considered that the H2O molecules which cause the hydrolysis are those of the solvent itself; but is clear on the assumption (which is discussed in the following chapter) that the water which enters into a colloidal complex is in multimolecular form, represented by the formula (H2O)n, in which the oxygen atoms are quadrivalent and, hence, much more active chemically than as illustrated in the simple solvent action of water.
Hence, the surface adsorption of reacting bodies by a colloidal catalyst may have a very important influence in decreasing the time required to bring the reacting molecules into intimate contact, and so increasing the velocity of the reaction.
But the colloidal condition of the catalyst may also aid in decreasing the "chemical resistance" which tends to slow up the reaction. Chemical resistance may be understood to be the internal molecular friction of the densely packed atoms within the reacting molecule, which tends to prevent the molecular rearrangement and so to prolong the second period of the reaction time. To overcome this friction and so decrease the reaction time, some form of energy is necessary. If there be present in the solution in which the reaction is taking place some colloidal catalyst, and if the reacting bodies are concentrated at the surface boundaries between the two phases of the colloidal system, they may be conceived to be within the sphere of influence of the surface energy of the dispersed particles of the catalyst, so that this may furnish the energy necessary to overcome the chemical resistance of the reacting bodies, and so to speed up the second portion of the reaction time.
From these considerations, it would appear that the colloidal condition of such catalysts as enzymes, etc., has much to do with their ability to increase reaction velocities, both by reducing the time necessary for the reacting bodies to come into molecular contact and by furnishing the energy to overcome the chemical resistance to the molecular rearrangement which constitutes the[Pg 218] reaction itself. Evidence in favor of the accuracy of this view of the nature of the catalytic action of colloidal substances is afforded by the facts that catalysts accelerate the velocity of reversible reactions in either direction and that they do not change the point of final equilibrium, in any case; that is, they do not affect the nature or direction of the reaction, but only accelerate a chemical change which would otherwise take place more slowly because of the stability (or chemical resistance) of the molecules involved, or their inability to come quickly into intimate molecular contact.
These facts and principles have been clearly established in many studies of the nature of enzyme action (enzymes are typical colloidal catalysts) and probably apply equally well to the action of other types of colloidal catalysts. On the other hand, the catalytic action of certain inorganic and non-colloidal substances, such as the action of acids in accelerating the hydrolysis of carbohydrates, etc., may be conceived to be due to chemical influences upon the internal molecular resistance, which are similar in their effects, but entirely different in their mechanism, from the physical effects of the surface boundary phenomena of the colloidal catalysts.
INDUSTRIAL APPLICATIONS OF COLLOIDAL PHENOMENA
Large numbers of industrial processes are based upon colloidal phenomena. Many of these processes were known and practiced long before the nature of the phenomenon itself was understood. But with the coming of the knowledge of the nature, causes, and possibilities of the control, of the colloidal condition of the materials involved, immense improvements in the economy of the process, or the quality of the end-products, have been worked out, in many cases. Many volumes of treatises concerning the industrial applications of colloidal phenomena have been written. Any discussion of these would be out of place here; but the following list of examples will serve to illustrate the immense importance of these matters both in industry and to the needs of everyday life: the tanning of leather; the dyeing of fabrics; vulcanizing rubber; mercerizing cotton; sizing textile fabrics; manufacture of mucilages and glues; manufacture of hardened casein goods; manufacture of celluloid; production of colloidal graphite for lubrication; the prevention of the smoke nuisance by electric[Pg 219] deposition; the purification of sewage; the manufacture of soaps; the manufacture of butter, cheese, and ice cream; fruit jellies, salad dressings, etc. This list could be extended to a great length, but is already long enough to emphasize the very great importance and practical value of colloidal phenomena in daily life.
NATURAL COLLOIDAL PHENOMENA
Many of the phenomena of nature are colloidal in character. These may be observed in the mineral, the animal, and the vegetable kingdoms. Here, again, a lengthy discussion of the nature of these phenomena would be out of place in this connection, and a few typical examples will serve to illustrate the general importance in nature of this property of matter.
In the soil, the following properties are easily recognizable as definite colloidal phenomena: water-holding capacity of clays, silts, loams, etc.; adsorption (or "fixation") of soluble plant foods so that they are not readily leached out of the soil by drainage; flocculation and deflocculation of clay, etc.
In the animal body; the contraction of muscles, the conveyance of nerve stimuli, etc., are undoubtedly accomplished by colloidal changes; and the existence of insoluble casein and fat in colloidal form in milk insures the proper nourishment of the young of nearly all species of animals.
In both plants and animals, as will be pointed out in the following chapter, practically all the vital activities of the cell protoplasm are definite manifestations of colloidal phenomena. Enzymes perform their catalytic functions by reason of their colloidal form. Proteins exist in colloidal form and are the seat of all vital functions. The regulation of the passage of materials into and out of the cell is governed by minute changes in the electrolyte concentration, etc., which produce enormous changes in the colloidal character of the protoplasm.
It is apparent, therefore, that the study of the colloidal condition of matter and of the properties arising out of it is of immense importance to the biochemist. No other single field is capable of yielding more fruitful results to the plant physiologist, in his studies of the response of plants to changes in their environment, or of the mechanism by which plants perform their internal functions.[Pg 220]
References
Bechhold, H., trans. by Bullowa, J. G. M.—"Colloids in Biology and Medicine," 463 pages, 54 figs., New York, 1919.
Burton, E. F.—"The Physical Properties of Colloidal Solutions," 200 pages, 18 figs., London, 1916.
Cassuto, L.—"Der Kolloide Zustand der Materie," 252 pages, 18 figs., Dresden and Leipzig, 1913.
Liesegang, R. E.—"Beiträge zu einer Kolloidchemie des Lebens," 144 pages, Dresden, 1909.
Ostwald, W., trans. by Fischer, M. H.—"Theoretical and Applied Colloid Chemistry," 218 pages, 43 figs., New York, 1911.
Ostwald, W., trans. by Fischer, M. H.—"A Handbook of Colloid-Chemistry," 278 pages, 60 figs., Philadelphia, 1915.
Taylor, W. W.—"The Chemistry of Colloids," 328 pages, 22 figs., New York, 1915.
Zigmondy, R., trans. by Alexander, J.—"Colloids and the Ultramicroscope," 238 pages, 2 plates, New York, 1909.
Zigmondy, R., trans. by Spear, E. B.—"The Chemistry of Colloids," 274 pages, 39 figs., New York, 1917.
CHAPTER XVI
THE PHYSICAL CHEMISTRY OF PROTOPLASM
Thus far, we have considered the chemical nature of the various groups of compounds which are found in the tissues of living organisms, laying emphasis upon those which are of plant origin. These compounds constitute the material, or machinery, of the cell, and their various transformations furnish the energy for its operation. We come now to a study of the mode of its operation, or the processes of vital phenomena.
Our knowledge of these matters is not yet far enough advanced to permit a definite statement as to whether there is any difference between the protoplasm of plant tissues and that of animal origin in their modes of action, or in the physical-chemical changes which constitute the vital phenomena in the two groups of living organisms. Thus far, no such differences have been discovered. Hence, in the following discussions, no attempt is made to differentiate between animal and plant protoplasm. Most of the facts and principles which are here presented have been developed as the result of the study of the physiological chemistry of animal life. No similar careful study of plant chemistry has yet been carried out; but preliminary studies seem to indicate that the same general principles apply to all protoplasm, regardless of whether it is of plant or of animal origin. It is possible, of course, that further studies of plant protoplasm will render necessary some modifications of some of these views as applied to the growth of plants; but they are believed to represent the best which is now known of the physical chemistry of the plant-cell activities.
HETEROGENEOUS STRUCTURE OF THE CELL
Examination of cell protoplasm under the microscope reveals that it is not a simple homogeneous mass. In the first place, it[Pg 222] has a definite structure, composed of (a) a nucleus; (b) numerous granular bodies of different sizes and kinds; and (c) a clear mass of colloidal material, which (if observed under the ultramicroscope, or photographed by ultra-violet light) is apparently made up of very minute particles of many different types of materials; the whole mass, in the case of plant protoplasm, being generally surrounded by (d) a differentiated layer known as the cell-wall. The actual internal structural arrangement of the clear colloidal mass is uncertain; but its properties indicate that it may be considered to be like a mass of foam (resembling a compact mass of soap-bubbles) the compartments of the foam being, of course, very minute and the films themselves almost infinitely thin, the contents of each compartment being probably liquid, and the whole composing a typical colloidal gel of complex composition.
This conception may not be accurate in every detail, but it seems to fit very closely the conditions and reactions of cell protoplasm. Furthermore, it is obvious that the definite structure, or form, of the cell is essential to its life; since, if the structure be destroyed by any kind of mechanical injury (freezing of the cell contents, resulting in the puncturing of the membranes by ice crystals; rupturing of the films, or cell-walls, by grinding with sharp sand, etc.) so as to bring about an intermingling of the parts which are segregated from each other in the organized structure, there results an immediate exhibition of abnormal chemical actions, accompanied by the liberation of carbon dioxide, and the death of the cell.
A proper mental picture of the organization of the cell structure and of the interrelation of all its working parts is suggested by the figure of a well-organized chemical factory, with the different chemical transformations which are involved in the whole process being carried on in different portions, or rooms, of the factory, with the various intermediate and final products regularly and systematically transported from one room to another as they are needed to keep each individual step in the whole process going at the proper rate, and with the different parts of the whole factory working in smooth coordination with each other. Any disturbance of the mechanism in any particular room, or any abnormal condition which breaks down the coordination or results in the mixing of the reagents or processes of adjoining rooms in improper order or proportions, produces instant destruction of the normal[Pg 223] process, abnormal reactions take place, and the factory output is interrupted.
No other conception than this one of a definite structure and coordination of the different working parts of a cell can adequately account for the great variety of chemical changes which are constantly going on in any given cell. It is wholly inconceivable that a homogeneous mass of all the varying chemical compounds which are contained in any given quantity of protoplasm could either exist or produce any regular sequence of chemical reactions. Structure, or organization of the cell-contents into separate colloidal compartments, and the segregation of cell-contents into masses having different functions, is essential to any reasonable conception of how the cell performs its various activities.
The best understanding of the structural arrangement is afforded by the conception that protoplasm consists of a colloidal gel, or sometimes a very viscid sol, containing water, salts, carbohydrates, fats, proteins, and enzymes. Evidence in favor of this conception is afforded by the appearance of protoplasm under a high-power microscope, and by the close resemblance of the processes which go on in it, and its responses to external stimuli, to those of an artificial gel of similar chemical composition.
Two different conceptions of the form in which the chemical components exist in this mass have been advanced. One is that they are in true molecular unions, known as "biogens," and that the reactions which take place in the mass may, therefore, be studied from the same basis as are reactions between similar substances when they take place in a beaker or test tube in the laboratory. It would seem, however, that the constantly varying proportions of the materials themselves, and the lack of homogeneity of cell contents, afford insurmountable difficulties to this conception as a basis for the study of cell activities. The other, and seemingly more reasonable, conception is that these bodies exist in the form of colloidal complexes, whose composition may vary within wide limits and whose reactions are responsive to the usual phenomena incident to the colloidal condition of matter.
According to the latter conception, vital activities of cell protoplasm may be due to changes in water content, to electrical disturbances, to the phenomena resulting from the conditions brought about by surface boundaries between the different phases of the gel, to varying osmotic pressure, to changes in chemical[Pg 224] reaction, etc., and may be controlled by various stimuli of chemical, physical, or mechanical nature. This conception seems, therefore, to fit most closely the actual conditions under which the protoplasm exists and carries on its vital functions.
With this conception in mind, we may now proceed to a consideration of how the various components of the complex organic colloidal system, and their specific properties, can affect its chemical activities.
The components of the system are, of course, water, salts, and the various organic compounds (fats, proteins, carbohydrates, and enzymes in all cells; and other groups, such as essential oils, tannins, pigments, etc., in cells which have certain special functions to perform) which constitute the solid phase of the colloidal mixture. In addition to the definite chemical properties of each of these component groups, which have been studied in detail in preceding chapters, there are many physical, or physical-chemical, properties of the system as a whole, and of its component parts, which are of the utmost importance in the physiological activities of the protoplasm. These we may now proceed to consider in some detail.
WATER
Water constitutes the largest proportion of the weight of active protoplasm. In living cell contents (except those of such bodies as resting seeds, etc.), water comprises from 70 to 95 per cent of the total weight of the substance; the average proportion being usually between 85 and 90 per cent. The fact that protoplasmic material can exist in turgid form with such high percentages of water as these is due, as has been pointed out, to its existence as a colloidal gel. It is because of this condition that increases in the proportion of water generally increase the turgidity, or turgor, of the protoplasm; instead of, as in all other cases, rendering the mixture less solid and more labile. Losses of water from the protoplasmic gel decrease its "swollen" condition and so render the tissue soft and flabby; while increases in water content swell the gel and make the tissue stiff and turgid. No other condition than that of a colloidal gel could respond in this way to changes in water content.
The formula which is commonly assigned to water is the sim[Pg 225]plest possible one; namely, H2O. But if the water were really as simple as this, the compound would boil at a very low temperature, would have a very low surface tension, etc.; whereas its actual boiling point, surface tension, etc., are much higher than those of other compounds having a higher molecular weight than is indicated by the formula H2O. Actual measurements of the physical properties of water indicate that at the temperature at which water is a vapor its formula is at least (H2O)2; while at lower temperatures, at which it exists as a liquid, its formula may be (H2O)3, or (H2O)4, or even more complex still. The cause for this association of the compound into multiple molecules undoubtedly lies in the extra valences of the oxygen. In many organic compounds oxygen is undoubtedly tetravalent, and it may be easily conceived that in these complex molecular groupings in the water it exhibits this same property; the possible molecular arrangements being represented by the formulas
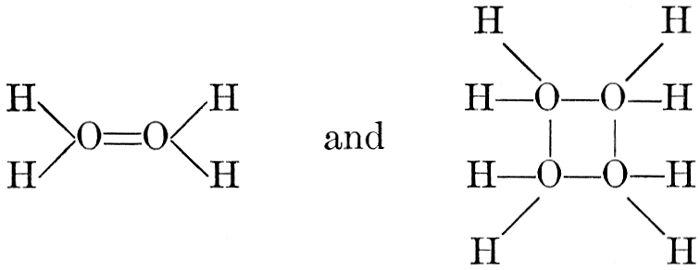
Such molecules may be conceived to break down very easily, leaving the extra valences of the oxygen available to form linkages with other atoms or molecules. This may constitute one of the ways in which water exerts its remarkable effects both as a solvent and as an accelerator of all kinds of chemical reactions. Other organic compounds which contain tetravalent oxygen are exceedingly active chemically, and there seems to be much to commend this view of the chemical structure of the water molecule.
Probably the most remarkable property of water is its power of solution. No other liquid surpasses water as a solvent. This power, as has been pointed out, is supposed to be due to, or in some way correlated with, the extra valences of the oxygen atoms, which may perhaps unite with similar extra valences of other substances with which the water is brought into contact, and so cause the latter to enter into solution. All kinds of substances dissolve in water, and when in solution, or even when only moistened, are much more active chemically than when dry. This property of[Pg 226] water contributes greatly to the possibilities of the chemical reactions which constitute life processes.
Water, likewise, has a higher dielectric constant than any other common liquid. This means that it does not readily conduct electricity, or readily permit electric equilibrium to be established in it; or, in other words, that it is a good insulator. This property permits the existence in it simultaneously of materials having opposite electric charges, or the so-called ionization phenomena; hence, water is the best-known ionizing medium, and ionization favors chemical reactivity.
Again, water has a very high specific heat, a fact which is of the utmost biological importance. It takes more heat to raise the temperature of one gram of water through one degree than is required to produce the same result in any other known substance; or, stated the other way around, a given amount of heat will cause less change in temperature of water than of any other known substance. Further, the latent heat of liquefaction and of vaporization (i.e., the amount of heat required to change the substance from solid to liquid and from liquid to gaseous state, respectively) is greater for water than for any other common substance. These facts are of very great importance in cell-protoplasm. The high specific heat of water provides that the heat liberated by the chemical reactions which take place in the protoplasm can be absorbed by the water of the cell contents, and given off again to other reactions, with very slight effect upon the temperature of the protoplasm itself. Hence, violent changes in temperature, which might be disastrous to the life of the cell, are prevented by the high specific heat of the water which it contains. Similarly, the high latent heat of liquefaction of water, resulting in the giving up of large quantities of heat before it can become solid, or "freeze," tends to prevent freezing and thawing of the cell contents with sudden changes of external temperatures at or near the freezing temperature of water.
As a result of its physical properties, as just briefly described, water accelerates all kinds of chemical reactions in protoplasm, both by solution and by ionization of such substances as undergo electric dissociation; and serves to regulate the temperature of the protoplasmic mass. Furthermore, in organic tissues, most of the important chemical reactions of the protoplasm are reversible hydrolyses; i.e., water actually enters into the reaction or is lib[Pg 227]erated by it, and the equilibrium point of the reaction is changed by the proportions of water which are present in the reacting mass. Hence, the presence of large proportions of water in the colloidal complex known as protoplasm has a very important influence upon its possibilities of biological reactions.
SALTS
Active protoplasm contains mineral salts in solution. These are of the same general nature as those found in sea-water, which is the original habitat of the earlier evolutionary forms of living matter. Or, it might be said that both plants and sea-water derive their mineral salts from the same source, namely the soluble salts of the soil. Recent investigations have shown that the proportions of sodium ions to calcium ions in sea-water are precisely those which maintain fats, proteins, etc., in a true colloidal emulsion; and that comparatively small variations in the ratio of these two cations produce very marked effects upon the colloidal conditions of these substances in an artificial colloidal preparation, which resemble very closely the changes which apparently take place in cell protoplasm under the influence of narcotics, or nerve stimulants, in blood-coagulation, in the parthogenetic development of germ cells, in cancerous growth of tissues, etc. In other words, in so far as it has been studied in this respect, cell plasma exhibits exactly the same responses to variations in the proportions of salts (electrolytes) in solution, that artificial emulsions of oils (fats) in water do; and the normal, or critical, equilibrium proportion of these electrolytes for all colloidal complexes is that in which they occur in sea-water. It must be admitted that there is as yet no definite evidence that the observations which have been made upon the protoplasm of animal tissues will apply equally well to plant cell protoplasm. But many of the phenomena which have been studied in animal tissues have what are apparently similar, if not identical, effects in plant tissues, and it seems reasonable to suppose that these conclusions apply generally to protoplasm of either animal or plant origin.
The effects which salts produce in protoplasm are undoubtedly due to the fact that, when in solution, they readily ionize and conduct the electric current. A discussion of the nature and importance of the theory of dissociation of electrolytes in solution,[Pg 228] or the so-called "ionization theory," which has done so much to clear up otherwise unexplainable properties of solutions, would be out of place here. But it may be noted that the ionized condition of salts in solution accounts for the avidity, or "strength," of acids and bases; for the increased osmotic pressure of such solutions; for the conduction of the electric current through solutions; and for the effects of these dissolved electrolytes upon the colloidal condition of many substances, since this is due to the electric charge on the dispersed particles.
Hence, the presence of salts in solution in the water of the protoplasm has a tremendous influence upon the osmotic pressure (which governs the movement of dissolved materials into and out of the cell protoplasm); upon the colloidal condition of the cell contents (which controls all the effects due to the surface boundary phenomena which are discussed below and which are responsible for a large part of the remarkable chemical activity of the protoplasm); upon the electrical phenomena (which constitute many of the stimulations which the protoplasm receives); and upon the acidity or alkalinity of the cell contents (which determine the nature of the respiratory, or oxidation, reactions of the protoplasm and, indirectly, its life or death).
The general nature of these physical-chemical properties of the protoplasm and of the relation of electrolytes in solution to them may now be considered in some detail.
OSMOTIC PRESSURE
Osmotic pressure is one of the chief factors in controlling the amount of water in the protoplasm. As is well known, the phenomenon known as "osmosis" is the passage of solvents, or of dissolved substances, into or out of any tissue, or substance, through the membrane which surrounds it. In the case of a cell, the membrane in question may be either the cell-wall or the internal colloidal films which are distributed throughout the entire mass of the cell contents.
From the standpoint of their relation to osmosis, membranes may be either impermeable, in which case neither solvent nor dissolved materials can pass through them; semi-permeable, which permit the passage of the solvent, but not that of dissolved crystalloidal substances; or permeable, which permit the free passage[Pg 229] through them of both solvents and solutes. The first and last of these types of membranes have no effect upon osmotic pressure; but osmotic pressure is at once set up whenever a semi-permeable membrane is interposed between solutions of different concentrations. It is due to the molecular motion of both the liquid and the dissolved solids, as a result of which a greater number of molecules are "bombarding," or pressing upon the membrane from the side of the more concentrated solution. This sets up an unequal pressure upon the two sides of the membrane, and if the latter be semi-permeable there will result a passage of the liquid through the membrane toward the denser solution so as to equalize the pressure. The resultant tendency is for the solutions on the two sides of the membranes to become equal in concentration by movement of the liquid from the less dense to the more dense portion, instead of by movement of the dissolved materials toward the less dense part of the solution as in the case of diffusion when solutions of different concentrations are brought in contact with no membrane to interfere with free diffusion.
Osmotic pressure tends, therefore, to force the movement of solvents through semi-permeable membranes from more dilute toward more concentrated solutions. Protoplasm acts in general as an approximately semi-permeable membrane or material. For example, if the concentration of sugar in any given mass of protoplasm becomes greater, by reason of the photosynthetic activity, osmotic pressure is set up and water enters the mass, thus preventing loss of turgidity due to increased concentration. Similarly, any other increase in concentration of synthetic products is compensated for by entrance of water because of increased osmotic pressure, unless the products are insoluble and, therefore, incapable of effecting the osmotic pressure.
Hence, osmotic pressure provides for the movement of water into and out of protoplasm and so tends to keep the proportion of water uniform throughout the entire tissue. It will at once occur to the reader, however, that if the statements in the preceding paragraph were unqualifiedly true, and if the protoplasmic mass were absolutely semi-permeable in character, there would be no possibility of the passage of dissolved solids into or out of the cell; i.e., if the protoplasm acted as an ideally semi-permeable membrane, only water could pass into or out of it. But we know that mineral salts from the soil must pass into any cell before the syn[Pg 230]thesis of proteins, etc., can proceed, and that the fats, carbohydrates, proteins, etc., which are synthetized in vegetative cells pass from these to other organs of the plant for use or storage. The obvious explanation for this condition of things in the plant is that protoplasm (and, indeed, this is equally true for practically all known membranes) is not absolutely impermeable to dissolved crystalloids; or, in other words, semi-permeability generally means only that the solvent passes through the membrane more readily and more rapidly than do the dissolved materials in it. Even colloidal materials will diffuse through most common membranes, although at so slow a rate that the process is scarcely observable by ordinary methods of study. Hence, the actual permeability of the protoplasm permits the movement of both water and dissolved solids from one part of the organism to another; but its approximation of semi-permeability produces osmotic pressure and induces freer movement of water than of dissolved substances, and so provides for turgidity of the cells and for equalization of the water content of different portions of the protoplasmic mass.
It is clear, therefore, that osmotic pressure plays an important part in the physical mechanism of cell activities and in the regulation of the proportion of water contained in the protoplasm, with its consequent effects upon the chemical reactions which may go on in the cell.
Actual measurements of the osmotic pressure of plant cell have been made. The results are more or less uncertain, because, as has been pointed out, a plant cell is not a definite quantity of uniform protoplasm surrounded by an ideal semi-permeable membrane, but is instead a mass of living matter which is approximately semi-permeable throughout its entire volume and is in a constantly changing condition because of the anabolic and catabolic activities which are going on in it; but values have been obtained which show a normal osmotic pressure as high as fourteen atmospheres in the cells of very turgid plants, such as those of some of the green algæ. Animal cells probably have an osmotic pressure similar to that of the blood which circulates around them, which is approximate that of seven atmospheres.[Pg 231]
SURFACE BOUNDARY PHENOMENA
In the preceding chapter, a brief consideration of the phenomena arising at surface boundaries was presented. It was pointed out that when any substance exists in the colloidal, or dispersed, condition, it has relatively enormous surface area and that, consequently, enormous surface boundaries between the dispersed phase and the dispersion medium exist in all colloidal mixtures. Since protoplasm is conceived to exist in the form of a colloidal gel, having a foamlike structure, it is apparent that it has these enormous surface boundaries between the different phases of the system, and that the phenomena arising from this condition are of great importance in its biological activities. The following necessarily brief discussion will serve to give some indication of the physiological importance of the surface boundaries in such a system.
It is easy to see that the molecules which are in the surface layers at the interface, where two phases of a colloidal system are in contact, are under the influence of forces quite different from those which are acting upon the molecules in the interior of either phase. It is apparent that the molecules in the surface layer are exposed on the inner side to the attraction and influence of similar molecules, while on the opposite, or outer, side they are exposed to the influence of molecules of an entirely different kind. This results in a state of tension, known as "surface tension," with the development of resultant forces and energy which profoundly affect the chemical reactivity of the molecules which are present in this surface layer. The so-called "surface energy," which results from this surface tension, produces marked increases in the possibility of chemical reaction between the materials which are present at the surface boundaries. In colloidal gels, this effect is so pronounced, in many cases, as to completely overshadow other types of influences upon reaction velocities. Also, the surface layer of a liquid is compressed by its surface tension, to such an extent that the solubility of substances in this surface layer is greatly increased over that of the same substances in the interior of the liquid, which results in greatly increased concentration of dissolved substances in the surface layer, and so increases the rate of chemical changes which take place there, as contrasted with the rate of the same reactions going on in the interior of the solution.[Pg 232] This latter consideration seems to be the factor of largest influence in colloidal catalysis.
But in addition to the increased rate of reaction in the surface layer due to the increased energy available there and to the increased concentration of dissolved substances, there is the possibility that the act of concentration itself bring into play molecular forces which give rise to a resultant increase in chemical potential, or chemical affinity, of the reacting materials, such as has been observed to result in other concentrated solutions. A discussion of the theoretical and mathematical considerations upon which this conception is based would be out of place here, but there is ample experimental evidence to indicate its soundness.
Further, as has been pointed out, colloidal phenomena are essentially due, in large part at least, to the electric charges on the dispersed particles. Electric charges accumulate at the surface of any charged body. Hence, the surface layers in any colloidal system carry its electric charges in highest concentration, and all of the chemical changes which are stimulated by electrical phenomena are most strongly influenced at the surface boundaries between the different phases of the system. This latter consideration affords a satisfactory explanation of the well-known depressing, or stimulating, action of electrolytes, especially acids and bases, upon the enzymic catalysis of protoplasmic reactions.
These few, brief statements are sufficient to indicate how extensively the chemical activities of colloidal protoplasm are influenced by the phenomena arising from the surface boundaries between different materials, which are present in such enormous extent in a colloidal gel. Surface boundary phenomena in a heterogeneous system, such as we have seen protoplasm to be, provide the possibilities for many reactions which would otherwise take place very slowly, if at all. Mere subdivision of the protoplasmic materials into the film, or foam, structure brings into play energies which may predominate over all other types of energy in the system. Here, too, effects may be extraordinarily modified by slight changes in environment, which effects could not be explained by any considerations which govern ordinary chemical reactions. Here, we deal with adsorption and other colloidal phenomena, rather than with ordinary stoichiometric combinations.
Indeed, it is not too much to say that the differences between[Pg 233] the chemical phenomena which are called "vital" and those which take place in ordinary laboratory reactions are due to the fact that the former are manifestations of the interchanges of energy between the different phases of a heterogeneous colloidal system, while the latter are governed by the laws of ordinary stoichiometric combinations.
ELECTRICAL PHENOMENA OF PROTOPLASM
The investigations of this phase of the physical chemistry of protoplasm have dealt almost exclusively with animal tissues and reactions, and have included the study of such phenomena as nerve impulses, muscular contractions, heart-beats, glandular secretions, etc. Tissues which respond to nerve, or brain, control are, of course, not found in plants. But there is plenty of experimental evidence to show that plant protoplasm carries electrical charges and exhibits electrical phenomena which are similar in character to those of animal tissues. In fact, it has been shown that the contraction of the lobes of the Venus' fly trap, when they close over an imprisoned insect, are accompanied by electrical phenomena in the leaf tissues which are precisely similar to those which take place in an animal muscle when it contracts. It seems probable that many of the observations and conclusions which have been derived from the study of the electrical disturbances in animal tissues may later be found to have definite applications to the vital phenomena of plant cells. Hence, it seems proper to give some brief consideration to these matters here.
The statement has been made that "every active living cell is essentially an electric battery," and it is believed that every activity of living matter, such as the rhythmic contraction of the heart, the passage of a nerve impulse, etc., is accompanied by an electric disturbance in the protoplasm of the tissues in question. Experimental proof of this electrical disturbance has been repeatedly obtained, by connecting a delicate galvanometer in a circuit through the living tissue which is undergoing different activities and obtaining widely varying readings of the instrument as the different phenomena are in progress, or by connecting the instrument with muscular tissue and observing its fluctuations with either the[Pg 234] irregular contractions of a voluntary muscle or with the rhythmic contractions of a heart muscle.
By means of such investigations as those just mentioned, it has been found that the part of the protoplasm which is most active is always electro-negative to the part which is less so; that is, the electric current flows from the more active to the less active portion of the protoplasm.
Many different explanations of the origin of the electric current which develops when the protoplasm is stimulated into activity have been suggested; but none of them have, as yet, any experimental confirmation. The most that can be said is that whenever any stimulus excites the protoplasm into activity, there is instantly developed in it an electrical disturbance, which continues as long as the action is in progress. Recent investigations, which have shown that there is a direct relation between many of the vital processes of protoplasm and the ratio of the electrolytes which it contains, particularly the ratio of sodium and potassium to calcium, would seem to indicate that the development of the electrical disturbance is a direct result of variations in the proportions of the salts of these metals, either brought about by, or themselves causing, changes in the permeability of the protoplasm, following the stimulus which determines the nature of the activity which it is to undergo. But there is as yet no indication concerning the mechanism by which this stimulation, with its resultant electrical phenomena, is transmitted to the protoplasm and accomplishes its characteristic effects.
ACIDITY OR ALKALINITY OF PROTOPLASM
The preceding sections of this chapter have dealt almost exclusively with the physical properties of protoplasm; including the phenomena of solution, ionization, surface boundary effects, and electrical disturbances, and their probable effects upon the chemical reactions which constitute its biological activities. It is necessary now to consider another phase of the physical chemistry of protoplasm, namely, its chemical reaction, whether acid, alkaline, or neutral, the effects of variation of this condition upon the activity of the protoplasm, and the mechanism by which it tends to preserve its own proper reaction in this respect.
The earlier methods of investigation of the chemical reac[Pg 235]tion of protoplasm were all based upon its color reactions to various staining agents. These sometimes led to erroneous conclusions, because of the effects of the staining agent itself upon the tissue; some stains are poisonous and result in the death of the protoplasm, others do not easily penetrate the semi-permeable colloidal mass, others are themselves changed by the oxidizing or reducing action of the protoplasm, etc. Again, colloidal adsorption effects often lead to the so-called "capillary segregation" of added staining materials. So that this method of study must be used with great care, or wholly erroneous conclusions will be reached, and many of the earlier reports have subsequently been found to be incorrect.
The recent improvements in the apparatus and methods for the determination of hydrogen-ion concentration have afforded a much more trustworthy method of determining the actual acidity or alkalinity of such materials than is obtained by color reactions, and this method is now being extensively used in the study of the reaction of active protoplasm.
It must be kept in mind that protoplasm is an heterogeneous mass and not an homogeneous solution, so that it is not always possible to determine the actual conditions as to neutrality of different parts of the protoplasm of a single cell, for example. Hence, one of the best methods of determining the reaction which is favorable to the life and activity of any given type of protoplasm is to investigate the reaction of a liquid medium in which the cells live and grow; this plan being based upon the assumption that a cell is not likely to have a reaction different from that of the medium which is favorable to its growth.
The results of all of the many investigations which have dealt with this problem point to the conclusion that the normal reaction for living protoplasm is either neutral or very faintly alkaline; but that it becomes acid when the cell is working in the absence of sufficient oxygen, and after the death of the cell.
The first effect of a change in the reaction toward acidity of the protoplasm is a decrease in the rate of respiration of the tissue, while increased alkalinity stimulates respiratory activity. Whet carried to the point of actual acidity, the respiratory coefficient becomes negative, and the cell actually gives off carbon dioxide because of the stoppage of the synthetic processes.
A second effect of change in reaction of protoplasm is to alter the enzymic activity of the cell. As has been pointed out, enzymes[Pg 236] are extraordinarily sensitive to minute changes in the reaction of the medium in which they are working. A change toward acidity in protoplasm immediately results in the stimulating of carbohydrate-splitting enzymes, which increases the supply of easily oxidizable simple carbohydrates, thereby tending to compensate for the decrease in respiratory activity. Further, increase in acidity increases proteolysis, thereby liberating alkaline ammonia-derivatives which tend to neutralize the rising acidity and so to restore normal neutrality or alkalinity. Thus it will be seen that in the very great sensitivity of its enzyme catalysts to slight changes in the reaction of the medium, the protoplasm possesses a very efficient mechanism for regulating changes and restoring equilibrium, if the latter be disturbed by any abnormal conditions. It should also be noted, at this point, that the almost universal presence in protoplasm of salts of carbonic and phosphoric acids acts as an additional "buffer" against pronounced changes in reaction of the material; the bicarbonates acting by means of their ready release or absorption of carbon dioxide, and the phosphates by their easy change from mono-sodium phosphate to di-sodium phosphate, and vice versa, the former being slightly acid and the latter slightly alkaline in reaction.
A third effect of increasing acidity is that it induces increased imbibition of water by the colloidal gel and causes swelling of the tissue. After death, when the reaction of the protoplasm becomes pronouncedly acid, this swelling often proceeds to the point of rupturing of the cell-wall, or internal membranes of the protoplasm, thus permitting the entrance of the putrefactive bacteria and hastening the decay of the tissue.
Finally, comparatively slight variations in the reaction of the protoplasm produce enormous changes in its colloidal condition, affecting in a very marked degree its permeability, its power of adsorption, etc.
It is clear, therefore, that variations in the chemical reaction of protoplasm profoundly affect its colloidal condition, its enzymic activity, and its respiratory processes. This necessarily brief survey is sufficient to indicate how important to the activity of the protoplasm is the chemical reaction of the material, and the mechanism with which it is provided for maintaining the favorable condition of neutrality or slight alkalinity.[Pg 237]
SUMMARY
It is evident that, within the limits of a single chapter, it has been possible to give only a very brief and incomplete discussion of some of the most important applications of the principles of physical chemistry to the properties and activities of protoplasm. Therefore, it may be profitable to summarize briefly these into a series of definite statements which may serve as a review of the principles which have been discussed in the preceding chapters, as applied to the activities of protoplasm.
Protoplasm is a complex hydrogel, composed of an heterogeneous mixture of proteins, fats, and carbohydrates, arranged in a foamlike structure, the compartments of the gel being filled with an aqueous solution of the soluble organic products of synthesis and of varying proportions of mineral salts which are of the same general nature as those of sea-water.
The gel is not uniform throughout the volume of any given cell, but is differentiated in different parts into what are known as the nucleus, the chloroplasts, the plasma of the cell, etc.
The vital activities of the cell consist in chemical reactions which are controlled by comparatively slight changes in the electrolyte distribution, or other environmental changes which affect the colloidal condition of the mass and, generally speaking, result in changes of the water content of the plasma, most such chemical changes being essentially reversible hydrolytic reactions.
The components of active protoplasm are in a condition most favorable to chemical reactions by reason of the enormous surface area of the colloidal material, resulting in abundance of available energy, intimate contact of the reacting materials, and the nearest possible approach to the condition of true solution which can be obtained without the loss of stable form and structure.
The reactions which take place in cell protoplasm, as a result of the action of either physical or chemical stimuli, are accompanied by electrical disturbances, which may be either caused by, or the result of, changes in the electrical charges of the mineral salts which are present in the gel. Such changes, like the chemical reactions which they accompany, may be regarded as reversible and mutually self-regulatory; so that the protoplasm has not only the possibilities of enormous chemical reactivity, but also the mechanism for self-regulation of its actions, the products or[Pg 238] results from any given series of changes generally tending to reverse the process by which they are proceeding and so to restore the condition of normal equilibrium.
Finally, the most characteristic difference between the reactions which go to make up the vital activities of a living cell and those of the same chemical substances when in inanimate form in the laboratory lies in the presence in the colloidal mass of the accelerating catalysts known as enzymes, which are produced by the protoplasm itself in some way which is as yet wholly unknown; and which not only add to the possibilities of rapid chemical change which are afforded by the colloidal nature of the material, but also, because of their extreme sensitiveness to minute changes in environmental conditions, serve to govern both the rate and the direction of the individual chemical reactions which constitute the vital activities of the protoplasmic mass. These enzymes are not distributed uniformly through any given cell, or organism, but are localized in different parts of the cell or tissue and so give to its different parts the ability to perform their various different functions.
References
Atkins, W. R. G.—"Some Recent Researches in Plant Physiology," 328 pages, 28 figs., London, 1916.
Czapek, F.—"Chemical Phenomena of Life," 152 pages, New York, 1911.
Czapek, F.—"Ueber eine Methode zur direkten Bestimmung der Oberflächenspannung der Plasmahaut von Pflanzen," 86 pages, 3 figs., Jena, 1912.
Höber, M. R.—"Physikalische Chemie der Zelle und der Gewebe," 671 pages, 55 figs., Leipzig, 1911.
Livingston, B. E.—"The Role of Diffusion and Osmotic Pressure in Plants," 149 pages, Chicago, 1903.
McClendon, J. F.—"Physical Chemistry of Vital Phenomena," 248 pages, Princeton University Press, 1917.
MacDougal, D. T.—"Hydration and Growth," Publication No. 297, Carnegie Institution of Washington, 176 pages, 52 figs., Washington, D. C., 1920.
Speigel, L., trans. by Luedeking, C. and Boylston, A. C.—"Chemical Constitution and Physiological Action," 155 pages, New York, 1915.
Thompson, D. A. W.—"On Growth and Form," 793 pages, 408 figs., Cambridge, 1917.
Willows, R. S. and Hatschek, E.—"Surface Tension and Surface Energy and their Influence on Chemical Phenomena," 116 pages, 21 figs., New York, 1919, (2d ed.).
CHAPTER XVII
HORMONES, AUXIMONES, VITAMINES, AND TOXINS
Reference has frequently been made, in preceding chapters, to the effect of various stimulating or inhibiting agencies upon the physiological activities of plant protoplasm. In the main, these agencies are external to the plant and are either physical, such as changes of temperature, amount of light received, etc.; or chemical, such as variations in the salts received from the soil, or common anæsthetics applied to the plants by man. A plant grows normally under certain conditions to which it has become adjusted by hereditary acquirements. When these conditions are altered, the effect upon the functioning of the plant protoplasm may be either stimulating or depressing. Extreme changes in environmental conditions generally result in the death of the plant; but changes which do not result in the lethal condition affect the plant by either stimulating it to more rapid physiological activity or by depressing its normal growth or functions. As has been pointed out, the same external influence, either chemical or physical, which acts as a stimulant if it differs only slightly from normal conditions, may become depressing, or positively toxic, if present to a larger extent.
There is also the possibility of the elaboration by the plant itself of internal agents, or substances, which may have a definite stimulating or inhibitory effect upon its metabolism and growth. The study which has been given to these matters has practically all been carried on within very recent years and is still in progress. Most of it is still in the experimental stage, in which no definite conclusions are as yet possible. Hence, the most that can be done at present is to give a brief review of the suggestions which have been made thus far, as indicative of the uncertainty of our present knowledge of these matters and of the general trend of the investigations which are now in progress.
Substances which are elaborated by plants and which are supposed to have a definite stimulating or beneficial effect upon[Pg 240] the activities of the plant which produces them, or to influence the physiological activities of other plants with which these substances come in contact through either the parasitic or the symbiotic relation, have been variously discussed under the names "hormones," "auximones," and "vitamines"; while injurious substances are generally known as "toxins." Whether these different terms actually represent different definite types of substances, or whether there are actually different groups of stimulating or inhibitory agents produced in plants, is uncertain; but the following brief statements will serve to indicate the general nature of the suggestions which have been put forward and of the experimental work which is now in progress.
HORMONES
The term "hormone" was first used to designate certain stimulating substances which are supposed to exist in the intestinal tracts of animals and to cause the glands to elaborate and secrete their characteristic enzymes. The supposed "hormones" are not themselves active in performing the digestive functions of the glandular secretions, but are the exciting, or stimulating, agents which cause the glands to secrete their active enzymes.
The same term has been used, by certain plant physiologists, to designate any agency, either external or internal, which stimulates plant protoplasm to abnormal activity. It has been pointed out that there are a variety of substances, which are themselves chemically neutral, that are powerful stimulants of vital activity if used in only minute proportions, but are powerful poisons if present in larger amounts. Many of the alkaloids act in this way upon the animal organism; while chloroform, toluene, and even some of the more complex hydrocarbons, act similarly upon the tissues of plants, and ether vapor is known to be a powerful stimulant in accelerating the flowering of plants and the ripening of fruits. It has been shown that the vapors of all such substances readily penetrate the protoplasm of leaves, seeds, etc., even when the same parts are impermeable to most mineral salts, sugars, etc.; and that upon entrance to the protoplasm of a leaf, or a seed, they tremendously stimulate its metabolic activity.[Pg 241] These hormones, as a class, are chemical substances which have very little attraction for, or power of combination with water; and it has been suggested that the ease with which they penetrate the protoplasm is due to the fact that they are not held at the surface by combination with the active water molecules which are present in the surface layer.
The principal effect which is supposed to be produced by these "hormones" is the stimulation of the enzymic activity, particularly that of the degenerative processes which take place late in the plant's life, at the flowering or ripening periods. Many of the changes which take place normally at ripening time, such as the change in color from green to yellow or red and finally to brown or black, when the fruit or vegetable is fully ripe, can be greatly accelerated by treatment with these substances. Hormones are similar in type to the ethereal salts, or esters, which constitute the natural essential oils that develop in many plants at this stage of their growth. Hence, it seems probable that these changes in plants which are maturing naturally may be hastened by the hormone action of the esters and similar bodies which are developed in largest quantities at that stage. It has been pointed out that the characteristic group which is present in many natural glucosides is of the same general type as the "hormone" substances which are used in the artificial stimulation of the flowering or ripening changes. This fact, together with the possibility of the liberation of greater percentages of these aromatic compounds from their glucoside combinations at the later periods of plant growth, is assumed, by some plant physiologists, to account for the change from synthetic to degenerative processes at this stage of the plant's development.
Further, it has been suggested that the autumnal coloration of leaves, and their dropping from the stems of the plant, as well as the ripening of seeds, is probably determined by the liberation in the plant, at that stage of its growth, or as a result of changed climatic conditions at that particular season of the year, of the hormones which either initiate or hasten the special enzymic changes which distinguish the degenerative from the synthetic processes of the plant.
Similarly, it has been suggested that parasitic fungi are able to penetrate the host plant by first excreting "hormones" which bring about degenerative changes in the tissues of the host plant[Pg 242] and so make it more easily penetrable by the hyphae of the parasite.
It will be seen that, in general, "hormones" are a type of substances (possibly often present in plants in the form of glucosides) which are supposed to stimulate the degenerative (or katabolic) vital processes in contrast to the synthetic (or anabolic) changes. It has been suggested that they do this in either one of two ways; namely, by favoring the introduction of water into the protoplasm and so diluting the cell contents, changing the osmotic pressure, etc.; or by bringing about a separation of the colloidal layers, or films, of the protoplasmic complex, producing a result similar to that produced by freezing the tissues. These ideas have been suggested by studies of the changes in the equilibrium of protoplasm when foreign substances are introduced into it. These studies have not as yet been brought to the stage of final conclusions, and the ideas presented must be considered as suggestive rather than as conclusive.
VITAMINES
"Vitamines," as contrasted with "hormones," are supposed stimulants of synthetic metabolic processes, or accelerators of growth, rather than of degenerative processes.
The term "vitamine" was first used to designate the substance, or substances, which must be present in the diet of animals in order that the animal organism may grow. Absence of these substances from the food of the animal results in the stoppage of growth of young animals and in various so-called "deficiency diseases" (such as beri-beri, scurvy, polyneuritis, etc.) of adults. This means that the animal organism is altogether unable to elaborate its own vitamines, and extended investigations have indicated that the vitamines necessary for animal uses are wholly of plant origin. The name "vitamine" was first used because it was supposed that these substances are chemical compounds of the amine type and, since they are necessary to normal life processes of animals, the name "vitamine" seemed to represent both their chemical character and their functions. Later investigations have caused doubt as to the accuracy of the first belief as to their chemical nature, and various other names have been suggested for the general group of substances which have the observed bene[Pg 243]ficial effects; while such specific names as "fat-soluble A," "water-soluble B," etc., have been used to designate individual types of these accessory food substances. However, the term vitamine is such a convenient one and is so generally recognized and accepted that it will probably continue to be used, at least until some more definite knowledge of the nature and composition of these growth-promoting, disease-preventing, and reproduction-stimulating food constituents is obtained.
The following definition of the term "vitamines" gives a satisfactory conception of the nature and functions of these substances, so far as they are yet known. "Vitamines; constitute a class of substances the individuals of which are necessary to the normal metabolism of certain living organisms, but which do not contribute to the mineral, nitrogen, or energy factors of the nutrition of those organisms." As sub-groups of the vitamines, there have already been recognized the growth-promoting, fat-soluble A; the antineuritic B, and the antiscorbutic C.
Until very recently, the investigations of vitamines have dealt exclusively with their relation to human nutrition; although it has been generally believed that the vitamines themselves are elaborated only by plants. It was generally recognized, however, that those plants, or parts of plants, which are capable of very rapid growth or metabolic changes, such as germs, spores, leaves, etc., are generally the richest source for vitamines for animal needs. Hence, there seemed to be considerable basis for the assumption that the elaboration of these substances by plants is definitely connected with their own metabolic needs. Recently, investigations of the functions of vitamines in the growth of plants have been begun. These are still in progress, but the following conclusions seem to be justified at the present time: (a) Potato tubers appear to contain growth-promoting substances which are essential to the proper growth of the sprouts. Whether these are the same substances which are efficient in the prevention of scurvy in men has not yet been investigated. (b) Baker's yeast is probably dependent upon a supply of vitamines in the medium in which it is to grow. Yeast itself, after having grown in barley wort, is one of the most important sources of vitamines for animal uses or for purposes of investigations of vitamine activity. But it has been reported that a yeast cell will not grow in an artificial medium which contains all the essential nutrients for yeast but[Pg 244] has no vitamines of other plant origin in it. The addition of barley wort, containing the vitamines from barley germs, or any other similar supply of vitamines, induces rapid growth and the storage of vitamines in the growing yeast masses. (c) The growth of many bacteria is either wholly dependent upon or greatly stimulated by the presence of vitamine-like substances in the medium upon which the microorganisms grow. (d) Sclerotinia cinerea, the brown rot fungus of peaches and plums, will grow only in a medium which contains, in addition to the essential sugar, salts, and nitrogenous material, vitamines derived from either the natural host plant tissues or other plant sources. These may be of two types (namely, a vegetative factor and a reproductive factor) or two different manifestations of activity of the same vitamine substance. But both of these factors must be provided before the fungus can make its characteristic growth.
There is, as yet, no conclusive evidence on many of the matters concerning the relation of vitamines to plant growth. But it seems that these substances are of almost universal occurrence in the organic world; that they are not of the same general type as other substances which are essential to the nutrition of plants or animals, but have specific stimulating or regulating effects upon the physiological activities of the organism; that the vitamines which are essential to animal life are elaborated by plant tissues, but that in the case of the bacilli of certain human diseases there seems to be some indication that the affected tissues of the animal host produce vitamines which are essential, or favorable, to the growth of the parasitic organism. There seems, therefore, to be evidence of a mutual relation between plants and animals with respect to their nutritional needs for the so-called "vitamines." But the evidence concerning the function of these substances in the tissues of the organism which elaborates them is, as yet, inadequate to provide any clear conception of the reason for their development or of the mechanism by which they are elaborated. Neither is there, as yet, any conclusive evidence concerning the chemical nature of the substances themselves.
AUXIMONES
Certain investigations have indicated that bacteria, at least, develop exogenous vitamines which are beneficial to the growth of[Pg 245] other plants. These are the so-called "auximones." For example, bacterized peat seems to contain auximones which may be isolated from the peat and exert a beneficial effect upon the growth of various seed-plants, including common farm crops. Neither the original experimental data, nor the theories which have been advanced to account for the observed beneficial effects of the supposed "auximones" have, as yet, sufficient confirmatory evidence definitely to establish their soundness. But it seems that there is a probability that some plants, at least, do elaborate vitamines, or auximones, which are useful to other plants.
TOXINS
Toxins are substances which affect injuriously the normal activities of the organism. As has been pointed out, they may be the same substances which, in lesser concentrations, exert a stimulating effect upon the same organism. Hence, it is probably inaccurate to discuss the toxins as a distinct group of substances.
There are, however, a large number of water-soluble chemical substances which are injurious to all living protoplasm, even at concentrations considerably less than the point of osmotic equilibrium in the juices of the protoplasm. These substances may act either directly or indirectly upon the protoplasm, but at certain concentrations they always affect it injuriously. In the main, these toxins are external agents of other than plant origin; although chemical substances developed by one plant may be toxic to other plants, or even to other organs of the same plant than those in which they are elaborated.
Toxins may be either general (i.e., injurious to all types of plants), or specific (i.e., injurious to only certain species) in their action. Examples of specific toxicity are of only minor importance in plant studies. They seem to be generally explainable on the basis of some unusual lack of resistance or failure of the susceptible plants to be able to exclude the entrance of these injurious substances into the protoplasm by "selective adsorption," or to convert the injurious substances into insoluble and non-injurious forms, as is done by other plants which are not susceptible to injury by these "specific" poisons. Hence, particular attention need not be given to this type of toxins.[Pg 246]
Toxic substances may act injuriously upon plant tissues in a variety of ways. Many electrolytes, especially the salts of the heavy metals of high valency, coagulate protein material and the entrance of such substances into the protoplasm causes disturbances in the colloidal condition which cannot be otherwise than injurious to its normal activities. Similarly, formaldehyde and many other organic compounds may affect the colloidal properties of the protoplasmic gel in such a way as to injure the plant tissues.
The same substance is sometimes much more injurious to the tissues of one part of a plant than it is to those of another part of the same plant. Thus, the rootlets of a young growing plant are much more susceptible to injury by many mineral salts than are the vegetative parts of the same plants; while anæsthetics of various kinds generally exhibit their greatest injurious effects upon the leaves, or synthetizing cells. Again, the mycelia of fungi are much more easily killed by toxic agents used as fungicides than are the spores of the same fungi. Some of these observed differences in toxicity may be due to differences in the physiological effect of the substance upon the protoplasm of the tissues which it enters, and others may be due to differences in the resistance of the protoplasm, or of its protective coverings, to penetration by the toxic material. Indeed, the possibilities of different types of toxic action, and of resistance to it by individual plants and species, are so varied that it is not possible to divide toxic agents into specific groups according to the nature of their injurious action upon the plant cell. They are, therefore, more commonly grouped into classes according to their chemical nature and economic significance as fungicides, as follows: inorganic and organic acids; caustic alkalies; salts of the heavy metals; hydrocarbon gases; formaldehyde; alcohols and anæsthetics; nitrogenous organic compounds; and miscellaneous decomposition productions of organic origin. The following brief review of some of the results of the experimental studies of the toxicity of different compounds belonging to these several groups will serve to indicate the general trend of the investigations of these matters which have thus far been made.
Acids.—The common inorganic acids (hydrochloric, nitric, and sulfuric) kill the rootlets of common farm crops when the latter are immersed for twenty to twenty-four hours in solutions of these acids containing from three to five parts per million of free acid.[Pg 247] Acetic acid must be about five times as concentrated as this, and other organic acids may be much more concentrated still before they produce the same injurious effects. The toxic effect of all these acids is greatly reduced in soil cultures, or if particles of sand, graphite, clay, filter paper, etc., are suspended in the solutions containing the acids, the reduction in toxic effect being probably due to the adsorption of the acids upon the solid particles. Hence, the concentrations which limit the toxic effects of these acids in water solutions cannot be taken as representing the condition with which the same plant will have to contend when growing under normal cultural conditions.
Alkalies.—The caustic alkalies must usually be present in from five to ten times as great concentrations as those of the mineral acids, in order to produce the same injurious effects upon the rootlets of common plants. The so-called "alkali" of soils is not alkali at all, but is neutral soluble salts present in sufficient concentration to exert a toxic effect.
Salts of the heavy metals are especially toxic to rootlets of plants. Salts of copper, mercury, and silver, have been found to kill the roots of seedlings immersed in them for twenty-four hours when present in proportions of less than three parts per ten million, while salts of many other heavy metals are toxic when present in concentrations of less than one part per million. The salts of the alkali metals are considerable less injurious than are those of the heavy metals, but even these exert their familiar injurious effect if present in concentrations which, measured by the ordinary standards, would still be regarded as very dilute solutions.
Illuminating gas, and similar hydrocarbon gases, kill plants when present in the atmosphere in as little as one part per million. Leaves, buds, and roots are all alike sensitive to this toxic effect, the nature of which is not yet understood.
Formalin, or formaldehyde, is a penetrating toxic agent for nearly all plant cells, and is commonly used as a fungicide for the destruction of parasitic fungi. It probably affects the colloidal condition in some way similar to its hardening effect upon gelatin, etc.
The toxic effect of many different organic compounds is so varied in its nature and extent that it is impossible to give any satisfactory brief review of its manifestations. Recent investigations appear to indicate that organic products of decomposition[Pg 248] of plant residues in the soil may exert powerfully toxic effects upon succeeding generations of the same, or of different, plants growing on the land. But the experimental data and conclusions concerning these matters are not yet accepted without question by all students of plant science or of the problems of the productivity of the soil. In fact, it is yet an open question whether toxic soil constituents are really an important factor in the so-called "unproductivity" of certain soils.
Alkaloids, and even the amino-acids which are produced in the tissues of some species of plants, while not toxic to the plants or organs which elaborate them, sometimes exhibit strikingly toxic action upon other plant organs with which they are brought into contact. There is, as yet, no satisfactory explanation of this difference in behavior between plant tissues toward various organic toxic substances.
In fact, the whole subject of the toxic action of various substances upon plants needs much more study before it is brought to the point where it will afford definite knowledge of either the physiological problems involved or of their practical applications in questions of soil productivity, etc.[Pg 249]
CHAPTER XVIII
ADAPTATIONS
Most of the discussions which have been presented in the preceding chapters have dealt with the types of compounds, the kinds of reactions, and the mechanism for the control of these, which are exhibited by plants under their normal conditions for development. The results of the evolutionary process have produced in the different species of plants certain fixed habits of growth and metabolism. So definitely fixed are these that in each particular species of plants each individual differs from other individuals, which are of the same age and have had the same nutritional advantages and environmental opportunities for growth, by scarcely perceptible variations, if at all. Indeed, this fixed habit of development makes possible the classification of plants into genera, species, etc. While different species of plants, given the same conditions of nutrition and environment, produce organs of the widest conceivable variety in form, color, and function; within the same species, the form and size of leaves, the position and branching of the stem, the color, size, and shape of the flower, the coloration and markings of the fruit, etc., are relatively constant and subject to only very slight modifications.
It is unnecessary to say that the mechanism, or the impulses, which govern the morphological characters of the tissues which any given species of plants will elaborate out of the crude food material which it receives from the soil and atmosphere, are wholly unknown to science. It is the commonly accepted assumption that the fixed habit of growth of the species is transmitted from generation to generation through the chromosomes of the germ cells. But the nature of the elements, or substances, which may be present in the chromosomes, which influence the character of the organs which will develop months later, after the plant which grows from the germ cell has gone through its various stages of vegetative growth, is still altogether unknown. There can be no question,[Pg 250] however, that some influence produces a fixity of habit of growth and development which is almost inevitable in its operation.
But while this unvarying habit of growth is one of the fixed laws of plant life, there are occasional deviations from it. A plant which, under normal conditions of growth, develops in a certain fixed way, when exposed to unusual environmental conditions, may, and often does, alter its habit of growth in what may metaphorically be said to be an attempt to adjust itself to the new conditions. Numerous examples of this phenomenon might be cited. Certain algæ, which grow normally in water at a temperature of 20° to 30° and which are killed if the temperature rises above 45°, have been grown for successive generations in water the temperature of which has been gradually raised, until they produce apparently normal growth in water the temperature of which is as high as 78°; also, certain types of algæ normally grow in the water of hot springs at temperatures of 85° to 90°, and others in arctic sea-water the temperature of which sometimes falls to -1.8° and never rises above 0° C. This phenomenon of the adjustment of a species of plants to new conditions, which in the case of farm crops is sometimes called "acclimatization," is of common occurrence and is often utilized to economic advantage in the introduction of new strains of crops into new agricultural districts. Again, the normal development of plants may be altered as the result of injury or mutilation. Thus, if the ear is removed from the stalk of Indian corn, at any time after flowering, there always results an abnormal storage of sucrose in the stalk, instead of the normal storage of starch in the kernels. Similarly, midsummer pruning of fruit trees generally results in the production of abnormally large number of fruit buds on the remaining limbs. Many other familiar examples of alteration of normal development in response to, or as the result of, abnormal conditions of growth might be cited.
TYPES OF ADAPTATIONS
To designate these different alterations of normal growth, several different terms have been used. Among these, "adaptation," "accommodation," and "adjustment" have been commonly used by different biologists. Sometimes these are used interchangeably, and sometimes different terms are used to desig[Pg 251]nate different types of response to altered conditions of growth. Inasmuch as there seems to be no generally accepted usage of these different terms, only one of them, namely, the word "adaptation" will be used here; and different manifestations of this phenomenon will be distinguished by using appropriate adjectives, as "physiological adaptations," "chromatic adaptations," "morphological adaptations," etc.
Two markedly different types of responses to altered conditions, or of adjustment to environment, may be recognized. In the first of these, for which we will use the term "physiological adaptation," the species of plant simply acquires the ability to exist and grow normally under conditions which formerly inhibited its growth. Thus, we may speak of the phenomena mentioned above as "acclimatization" as the physiological adaptation of the crop to the new conditions of growth. In general, physiological adaptations include such variations in the characters or habits of growth of plants as results in differences in resistance to heat or to cold, relations to water, aggressiveness in competition with other plants, etc. In such cases, no modification of the morphological characters of the plant can be observed, the changes which take place in the structure of the plant (if, indeed, there be any such changes) must be only minor adjustments of the protoplasm to meet the new environmental needs.
In the second type of adaptations, for which we will use the term "morphological adaptations," the structure, or color, or some other morphological character of the plant is actually changed in some easily recognizable way, in order that the plant may be better adjusted to its environment. As examples of morphological adaptations, there may be cited the change in color of sea-weeds with increasing depth in the sea, and other examples of chromatic adaptation which are discussed below; the development of fewer, or a larger number, of buds on the above-ground stems of plants, in response to decreases, or increases, in the available supply of food; the alteration in the size and shape of the leaves of many plants when they are grown in shade; the dwarfing of plants at high altitudes, or under conditions of severe drought; the development of underground storage organs for certain species of shrubs and trees which grow in regions that are subject to periodical burning-over, in such a way as to destroy the above-ground storage stems, etc.[Pg 252]
Hence, the two terms, as we will use them here, may be defined as follows: morphological adaptation is a change in the structural character of the species in order that it may be better fitted to meet the needs of the new conditions of growth; while physiological adaptation is an acquired power to survive and develop under abnormal conditions, which is not accompanied by any visible change in the characteristic structure of the species.
Both of these types of adjustment may be either hereditary (or evolutionary), or spontaneous in their origin and development. Changes which are evolutionary are fixed by heredity and become definite habits of growth in the species. Their origin may be explained in either one of two ways; namely, the so-called "increase by use," and "the survival of the fittest." The hypothesis of "increase by use," as an explanation of adaptations, is based upon the well-known observation that, in animals, muscles and other organs increase in volume as they are extensively used; and the assumption of the application of this principle to the phenomenon of adaptation supposes that the modification of any given structure or composition is the result of the hereditary accumulations of increased size resulting from use, or of atrophy from disuse. The "survival of the fittest" theory supposes that individuals of a species differ from each other by spontaneous variations, and that in the competitive struggle for existence those forms which are best adapted to the environmental conditions survive while the others perish. The contrast between these two views is that the first holds that adaptation proceeds by development, and the second that it proceeds by variation and elimination; the first presupposes the existence in the organism of a mechanism for response to changing conditions, and the second assumes that there are chance variations followed by the death through competition of the forms which are not able to meet the needs of the environment.
Confusion arises whenever an attempt is made to apply either of these theories to all kinds of adaptations. The idea of increase by use can be applied with some satisfaction to certain morphological adaptations in animal structure; and to such phenomena as the increase in strength of the branches of fruit trees, either with or without corresponding increase in size, as the load of fruit increases. But it certainly cannot apply to color change in surface pigmentation of either animals or plants, which is one of the most common[Pg 253] forms of adaptation. Furthermore, it is difficult to conceive the general application of this idea to alterations of habits of growth of plants, since a plant cannot have any such thing as a voluntary control over the amount of "use" which it makes of its different organs in response to changes of environment. The common form of statement that a plant develops an organ, or a process to meet a certain need, or modifies its habits of growth to meet a change of environment are, of course, purely metaphorical, and can only be taken to mean that such processes are mechanical responses to changes in external conditions.
The nature of the mechanism by which these responses are accomplished is, as yet, wholly unknown. There is accumulating a large mass of experimental evidence which goes to show that, while both temperature and light are very important factors in determining the type of changes which will take place in a living organism, the so-called "photochemical action of light" is by far the most potent of all the climatic factors which influence the course of development of a plant. But we have, as yet, no inkling of how the protoplasm of the plant adjusts or controls its responses to variations in any of these external factors.
With these general considerations in mind, we may now proceed to the consideration of certain particular types of adaptations.
CHROMATIC ADAPTATIONS
Adaptations have been observed in both the energy-absorbing pigments of the general tissues and in the ornamental epidermis pigments of plants. The former are by far the most important from the physiological point of view; while the latter may have interesting biological significance.
Under nearly all conditions of growth of land plants, the supply of the chlorophylls and their associated pigments provides for the absorption of solar energy far in excess of the amount necessary for the photosynthetic assimilation of all the carbon dioxide which is available to the plant. It has been shown that an active green leaf, on an August day, can absorb eight times as much radiant energy as would be required to assimilate all the carbon dioxide present in the air over its surface. No land plant, under normal conditions, develops supplementary pigments in[Pg 254] order to utilize other than the parts of the spectrum which are absorbed by chlorophyll and its associated pigments.
But deep-sea plants show quite a different phenomenon of pigment development. Water is a blue liquid. At depths of 40 feet or more, the light which penetrates is devoid of red rays, feeble in yellow, and is characteristically green or blue in color. Now, the red rays of the spectrum are the ones which are most efficient for photosynthesis. Sea weeds which grow at these depths are brilliantly red in color, at intermediate depths they are brown, and at the surface they are green, in the same latitudes. While it is possible that the temperature of the water at these different depths may have something to do with the chemical synthesis of the pigments, it appears plain that this color change at increasing depths is a definite adaptation to provide for the absorption of the solar energy which is available at these depths. It has been shown that these pigments of deep-sea plants are additional to, and not substitutes for, the chlorophylls, etc. The latter pigments are present in normal amounts, but are supplemented by those which absorb the green and blue portion of the spectrum. Hence, this type of adaptation might be conceived to be a "survival of the fittest," resulting in the "natural selection" of individuals of the highest total pigmentation. But, on the other hand, there is experimental evidence to show that plants possess some means of varying their pigmentation in response to the character of the light which comes to them. For, it has been found that a complete change in color of certain highly colored plants can be produced in a single generation, by growing the plants in boxes or chambers whose walls are composed entirely of differently colored glass, so that the plants within receive light of only a particular part of the spectrum. In such cases, the plant, starting with an initial "natural" color, changes through a succession of colors until it finally reaches equilibrium at one which provides for the proper absorption of the right kind of light from the new supply which is available to it. Hence, it seems proper to conclude that chromatic adaptation is not a process of "natural selection," but a definite result of an actual mechanism for adaptation to changed environmental conditions of supply of radiant energy.[Pg 255]
STRUCTURAL ADAPTATIONS
Changes in structure to meet special conditions of growth may be of several different types.
One of these, which is often cited as an example of adaptation (in this case, the term is used with a significance quite different than that in which it is being used here) is that of the development of unusual and often fantastic shapes of flowers, which are so related to the anatomy of certain species of insects that visit these flowers in search of nectar, that provision for the cross-fertilization of the plants is insured, in that the pollen from the anthers of one flower becomes lodged on the body of the insect as it is withdrawing from the flower in such a way that it comes in contact with the pistil of a second flower as the insect enters it. Such flowers often have such peculiar shapes and lengths of nectar tubes, etc., that only a single species of insect, whose anatomical shape is "adapted" to that particular blossom shape can enter the flower in its search for nectar. It is clear that this form of "morphological adaptation" is a highly specialized one, which can only be the result of a long process of evolutionary development. It is obvious that the plant cannot possibly possess a mechanism, or ability, to alter its flower form in order to make it conform to the shape and length of the proboscis, or other body parts, of a particular species of insect. Either the insect or the plant, or both, must go through a process of evolutionary development in order to arrive at this form of mutual "adaptation."
A form of true morphological adaptation (in the sense in which we have been using the term) is exhibited by many species of plants, which are provided with many more buds, or growing points, than ever actually begin to grow. For example, the single plumule which develops from a germinating wheat embryo has at its upper end a hundred or more tiny growing points. At the proper stage of its growth, several of these tiny buds begin to grow into individual separate stems, and the new wheat plant thus produces several stems from one seed and root system, a process known as the "stooling." The number of stems in a single "stool" depends upon the number of the potential growing points which are stimulated into growth. It varies from only two or three up to as many as thirty or forty, and is apparently controlled by the favorable or unfavorable conditions of climate or[Pg 256] nutrition at the time when the "stooling" takes place. The plant is thus provided with a mechanism for adapting its possibilities of growth to the supply of growth-promoting material which is available to it.
Many other plants produce far more buds than ever develop into growing tissues, and buds which, under normal conditions, remain dormant, under altered conditions start into growth and so provide for an "adaptation" of the total mass of the growing plant to correspond with the altered conditions of growth. The actual means by which certain buds are stimulated into growth while others remain dormant, or are inhibited from growing, are as yet unknown. Two theories have been advanced. One is that the growing buds absorb all available nutrition and the others remain dormant by reason of lack of growth-promoting material. The other is that the vegetating (growing) tissue elaborates and sends to other parts of the organism one or more substances, which actually inhibit growth of the other parts, as dormant buds, etc. The experimental evidence which has been presented thus far is inconclusive, but seems to favor the distribution of nutritional material as the governing factor, although there is some evidence which seems to indicate that a supposed growth-inhibiting substance is actually translocated from rapidly-vegetating tissues to other parts of the plant. There is, however, no explanation of how the buds, or other tissues, which do grow get their initial stimulus, while the dormant buds do not. After growth has once started, the changes in osmotic pressure due to the accumulation and translocation of synthetized materials can account for the movement of new nutritional material for the synthetic processes into the growing organ; but this would not account for the selective stimulation of only a part of the buds, or possible growing points, of a plant, or for an adaptational development of others under altered conditions of growth.
The form of morphological adaptation which has been discovered in the course of the study of the native vegetation of the campos of Brazil (which have a very dry season and have been regularly burned over by the natives for many generations) in which the papilionaceous shrubs have developed underground trunks, or stems, and seem actually to profit in luxuriance of growth when the rainy season comes on by reason of this morphological adaptation to the unusual environmental conditions,[Pg 257] is wholly inexplicable by any present knowledge of the science of plant growth.
PHYSIOLOGICAL ADAPTATIONS
The type of adjustment to environmental conditions which does not result in any recognizable alteration in the structure of the plant, but simply permits it to grow under new conditions, manifests itself in many ways. These adjustments are usually associated with differences in temperature during the growing season, and for this reason, most such examples of adaptation have been studied in connection with possible temperature reactions upon the growing organism.
However, recent investigations seem to point strongly to the conclusion that the amount of light rather than the temperature of the new surroundings is the most important influence in determining the physiological processes known as the "acclimatization" of plants. For example, a very elaborate series of investigations has shown that the flowering stage in the development of plants is determined by the length of the daylight period per day, irrespective of the actual amount of vegetative growth which the plant has made. Thus, tobacco plants, which during a period of long days grow to the height of 8 or 10 feet before blossoming, if grown at the same temperature in periods of short days (or if kept in the dark during a portion of the longer days) will blossom when less than 3 feet in height and when the total mass of vegetative material which has been produced is less than one-third of that of the "gigantic" plants of the same variety grown with longer periods of illumination per day. This same principle has been found to hold good for many widely different types of plants. In some species, however, flowering is favored by long days, and vegetative growth by short daylight illumination. But in all species which have been studied, there seems to be a direct relation between the length of day, or the total illumination per day, and the normal or abnormal functioning of the plant. It is apparent that at least the physiological function of sexual reproduction (flowering and seed-production) is determined by the length of daylight illumination. The duration of daylight per day which is necessary to induce the blossoming of the plants varies for different species, but it is constant for individuals of the same[Pg 258] species. This adaptation of stage of growth to duration of daily illumination must, therefore, be an evolutionary character of the species.
Hence, it appears that in many cases physiological adaptation may be a direct response of the life-processes of the plant to the daily length of photochemical stimulation which it receives from solar light. But there is, as yet, no explanation of how this (or any other) influence actually changes the vital processes of the plant protoplasm so as to bring about either a morphological adaptation of structure or a physiological adaptation of functions to altered conditions of growth.
CONCLUDING STATEMENTS
Enough has been said to show how very inconclusive and unsatisfactory is our knowledge of the phenomena known as "adaptation." Even the nomenclature used by different scientists to describe its various manifestations is confused and misleading. For example, certain crops are said to be "adapted" (i.e., suited) to certain types of soils, and vice versa; crops are said to be "adapted" to given agricultural districts, etc.
In this chapter, an attempt has been made to arrange in some semblance of order some of the known manifestations of alteration of fixed habits of growth of plants in response to changes of environment, and to point out some of the suggestions of possible explanations of these phenomena which have been presented by different investigators.
This presentation cannot be considered as anything other than an introduction to a field of study which is as yet almost entirely unexplored, and, like all other unexplored territory, is full of mysteries. If the study of this chapter serves to stimulate interest in these mysteries and wonders of plant life, its purpose will have been accomplished.
INDEX
Bold-face figures indicate main references
- Accelerators, 196.
- Accessory substances, 19.
- Achroo-dextrin, 61.
- Acid, acetic, 125, 126, 128, 132, 133, 136.
- arabic, 98.
- arachidic, 133.
- aspartic, 168, 177.
- brassic, 133.
- butyric, 126, 133.
- capric, 133.
- caprylic, 133.
- carnaubic, 140.
- cerotic, 133, 140.
- citric, 125, 127.
- convolvulinic, 81.
- crotonic, 133.
- diamino-oxysebacic, 169.
- diamino-trioxydodecanic, 169.
- digallic, 96.
- ellagic, 96.
- euxanthic, 84.
- formic, 25, 126, 128, 132.
- galactonic, 42.
- gallic, 96.
- geddic, 69.
- gluconic, 42.
- glucuronic, 42, 43.
- glutamic, 168, 177.
- glycero-phosphoric, 142.
- hydrocyanic, 77.
- jalapinic, 81.
- lauric, 133.
- lignoceric, 133.
- linoleic, 133.
- linolenic, 133.
- malic, 124, 127.
- malonic, 124.
- mannonic, 42.
- melissic, 133.
- meta-pectic, 68, 70.
- mucic, 68.
- myristic, 133.
- nitric, 125.
- nucleic, 162.
- oleic, 133.
- oxalicic, 68, 124, 125, 126, 128.
- palmitic, 133, 140.
- parapectic, 70.
- pectic, 70.
- phosphoric, 141, 142, 162.
- propionic, 126, 166.
- pyrocatechuic, 96.
- quercitannic, 98.
- racemic, 54.
- ricinoleic, 133.
- ruberythic, 83.
- saccharic, 42, 68.
- salicylic, 81.
- sarco-lactic, 128.
- stearic, 131, 133.
- succinic, 127, 128.
- sulfuric, 125.
- sylvinic, 149.
- talonic, 42.
- tannic, 97, 127.
- tartaric, 127.
- uric, 160.
- xanthoproteic, 173.
- Acid amides, 151.
- Acidity of protoplasm, 234.
- Acid glucosides, 81.
- Acid potassium oxalate, 125.
- [Pg 260]Acid potassium sulfate, 88.
- Acid salts, 124.
- Acids as toxins, 246.
- Acid sodium sulfate, 125.
- Acrolein, 135.
- Acrose, 28.
- Activators, 196.
- Adamkiewicz's reaction, 173.
- Adaptations, 249.
- Adenase, 190.
- Adenine, 160, 162.
- Adipo-celluloses, 74.
- Adsorption, 214.
- Æsculetin, 81, 82.
- Æsculin, 81, 82.
- Ætiophyllin, 106, 107, 109.
- Ætioporphyrin, 108, 109, 110.
- Alanine, 168, 177.
- Albumins, 175, 176.
- Albuminoids, 175, 176.
- Alcogel, 205.
- Alcohol, ethyl, 40, 125,
- Alcohol glucosides, 80.
- Alcosol, 80.
- Aldehyde, benzoic, 148..
- cinnamic, 148.
- formic (see formaldehyde).
- glyceric, 35.
- Aldehyde glucosides, 80.
- Aldehydrol, 46.
- Aldonic acids, 42, 44.
- Aldose, 32.
- Alizarin glucosides, 78.
- Alkalinity of protoplasm, 234.
- Alkalies as toxins, 247.
- "Alkali salts," 10, 247.
- "Alkali soils," 10, 14.
- Alkaloidal reagents, 154, 172.
- Alkaloids, 18, 20. 151, 153, 248.
- Allose, 36, 37.
- Allyl isosulfocyanide, 88, 89, 148.
- Allyl sulfide, 148.
- α-glucose, 46.
- α-glucosides, 55.
- α-methyl glucoside, 47.
- Altrose, 36, 37.
- Aluminium, 4.
- Amandin, 170, 176.
- Amines, 151.
- Amino-acids, 6, 151, 166, 179, 248.
- Ammonia, 152.
- Ammonium hydroxide, 142, 152.
- Ammonium salts, 6.
- Amorphous chlorophyll, 104, 105.
- Amphoteric electrolytes, 172.
- Amygdalase, 87.
- Amygdalin, 81, 86.
- Amyl acetate, 148.
- Amylase, 186, 189, 191.
- Amylo-cellulose, 60.
- Amylo-dextrin, 61.
- Amylo-pectin, 60.
- Amylose, 60.
- Anergic food, 2, 17.
- Animal nucleic acids, 162.
- Antagonism, 14.
- Anthocyans, 83, 102, 115, 121.
- Anthocyanidins, 116.
- Anthocyanins, 102.
- Anthoxanthins, 117.
- Anthraquinone, 83.
- Antienzymes, 120, 197, 198.
- Antioxidase, 120.
- Antiscorbutic C, 243.
- Apigenin, 84, 118.
- Apiin, 84.
- Apiose, 84.
- Araban, 69.
- Arabinose, 35, 44, 68, 69, 88.
- Arabinosides, 56.
- Arbutin, 77, 79,
- Arginine, 169, 171, 177.
- Arsenic, 13.
- Asymmetric carbon atom, 33.
- Atropine, 155, 156.
- Autotrophic plants, 16, 18.
- Auximones, 239, 240, 244.
- [Pg 261]Available plant food, 4.
Baptigenin, 79.- Baptisin, 79.
- Beeswax, 133.
- Beet sugar (see sucrose.)
- Berberine, 155.
- Betaine, 152.
- β-glucase, 55.
- β-glucose, 46.
- β-glucosides, 55.
- β-methyl glucoside, 47.
- Biogens, 223.
- Biological significance, 19.
- Biuret reaction, 173.
- Borneol, 148.
- Boron, 13.
- Bromelin, 189.
- Brucine, 157.
- Buffers, 236.
- Butter fat, 133.
- Butyric acid ferment, 190.
Cadaverine, 152.- Caffeine, 160.
- Calcifuges, 9.
- Calciphiles, 9.
- Calcium, 3, 5, 9, 10, 14, 68.
- Calcium oxalate, 126.
- Campferitrin, 118.
- Campferol, 118.
- Camphene, 147.
- Camphor, 148.
- Cane sugar (see sucrose.)
- Caoutchouc, 147.
- Capillary segregation, 235.
- Carbohydrases, 189.
- Carbohydrates, 18, 20, 21, 30, 163, 234.
- Carbon dioxide, 2, 3, 18, 21, 22, 23, 24, 40, 222.
- Carbonic acid, 227.
- Carbon monoxide, 24.
- Carboxyl, 124.
- Carboxylases, 186, 190.
- Carnauba wax, 133, 140.
- Carotin, 112, 113, 121.
- Carnauba wax, 102, 111.
- Carvacrol, 148.
- Casein, 165.
- Castanin, 176.
- Castor oil, 130.
- Catalases, 190, 193.
- Catalysis, 182.
- Catalysts, 17, 25, 183.
- Catechol tannins, 97.
- Catechin, 97.
- Catechu tannins, 97.
- Cellobiose, 52.
- Cell structure, 221.
- Cellulase, 71, 186, 189.
- Celluloid, 52.
- Cellulose, 20, 45, 63, 67, 72.
- Cell-wall, 9, 12, 222.
- Cerebrosides, 141, 144.
- Chemical resistance, 52.
- Cherry gum, 68.
- Chinovose, 35.
- Chlorine, 12.
- Chlorophyll, 10, 11, 21, 27, 102, 105, 110, 111, 113, 122, 254.
- Chlorophyll a, 103, 106, 107, 111.
- Chlorophyll b, 103, 106, 108, 111.
- Chlorophyllase, 104.
- Chlorophyllin a, 106, 107.
- Chlorophyllin b, 106, 107.
- Cholesterol, 129, 136.
- Choline, 89, 103, 141, 142, 152.
- Chromatic adaptations, 251, 253.
- Chromogens b, 92, 119.
- Chromo-proteins, 175.
- Chrysin, 117.
- Cinchonine, 155, 157.
- Coagulated proteins, 175.
- Coagulation enzymes, 190.
- Cocaine, 155, 157.
- Cocoanut oil, 133.
- Codeine, 155, 157.
- Coenzymes, 197.
- "Cold-drawn oils, 137.
- Collodion, 73.
- Colloidal phenomena, 17, 202.
- Colloidal solutions, 204.
- Colloids, 202.
- Colophene, 147.
- [Pg 262]Colophony, 149.
- Compound celluloses, 71, 73.
- Conglutin, 176.
- Coniferin, 80.
- Coniine, 155, 156.
- Coniine, 165, 174, 175.
- Continuous phase, 203.
- Convolvulin, 81.
- Copper, 13, 247.
- Cork tissue, 99, 101.
- Corn oil, 130.
- Corylin, 176.
- Cottonseed oil, 130.
- Critical elements, 4.
- "Crude fat," 141.
- Crystalline chlorophyll, 104, 105.
- Crystalloids, 202.
- Crystalline chlorophyll, 81, 148.
- Curarine, 157.
- Cuto-celluloses, 74.
- Cyanidin, 85, 116.
- Cyanin, 85.
- Cyanophore glucosides, 86.
- Cyanophyllin, 107, 108.
- Cyanoporphyrin, 108.
- Cymarigenin, 90.
- Cymarin, 90.
- Cymarose, 90.
- Cystine, 168, 171.
- Cytase, 107, 108.
- Cytosine, 161, 162.
Daphnetin, 81. 82.- Daphnin, 81.
- Deaminases, 168, 190.
- Delphinidin, 85, 116.
- Delphinin, 85.
- Derived proteins, 173, 175, 177.
- Dextrin, 59, 61.
- Dextrinase, 189.
- d-galactose, 33.
- d-glucose, 33.
- Dextrosans, 59.
- Dextrose (see glucose.)
- Dhurrin, 87.
- Diastase (see amylase.)
- Diastase of secretion, 191.
- Digitaligenin, 89.
- Digitalin, 89.
- Digitogenin, 89.
- Digitonin, 89, 90.
- Digito-saponin, 90.
- Digitoxigenin, 89.
- Digitoxin, 89.
- Digitoxose, 89.
- Diglycerides, 131.
- Diose, 30.
- Disaccharides, 31, 48.
- Dispersed phase, 203.
- Dispersion medium, 203.
- Dispersion phenomena, 203.
- Drying oils, 132.
- Dulcitol series, 36.
Edestin, 170, 176.- Egg-albumin, 165.
- Electrical phenomena of protoplasm, 233.
- Electrolytes, 213, 227.
- Emulsoids, 206, 214.
- Emulsions, 206.
- Emulsin, 55, 77, 87, 184, 189.
- Enol, 44, 56.
- Enzymes, 18, 18, 19, 20, 23, 26, 120, 121, 181, 183, 194, 199, 244.
- Erepsin, 189.
- Erythro-dextrin, 61.
- Erythrophyllin, 107.
- Erythrose, 35.
- Essential elements, 4.
- Essential oils, 18, 146, 147, 224.
- Esterases, 186, 189.
- Esters, 124, 125, 129.
- "Ether extract," 141.
- Etherial salts (see esters.)
- Ethersol, 205.
- Ethyl acetate, 125.
- Ethyl nitrate, 125.
- Excelsin, 176.
- Extracellular enzymes, 184.
Fats, 18, 20, 129, 224, 227.- Fat-soluble A, 243.
- Fatty acids, 132, 142.
- [Pg 263]Fehling's solution, 39, 47.
- Fermentability, 40.
- Ferments (see enzymes.)
- Ferric salts, 11.
- Ferrous salts, 11.
- Fisetin, 118.
- Flavone, 82, 83, 102.
- Flavonol, 84.
- Food, 1.
- Formaldehyde, 22, 23, 25, 26, 27, 247.
- Frame-work material, 20, 67.
- Fraxetin, 82.
- Fraxin, 82.
- Fructose, 23, 28, 32, 36, 41, 44, 45, 47, 57, 162.
- Fructosides, 41, 42.
- Fruit sugar (see fructose.)
- Fucose, 35.
- Fucoxanthin, 102, 112, 114.
Galactans, 47, 59, 63, 72.- Galactoheptose, 36.
- Galactooctose, 36.
- Galactose, 32, 36, 38, 45, 47, 57, 72, 77.
- Galactosides, 41, 42.
- Gaultherin, 81.
- Gel, 175, 205, 208.
- Gelation, 210.
- Gel-formation, 208, 211.
- Gentianose, 52, 53.
- Gentiobiose, 49, 52, 53.
- Gentisin, 119.
- Gitaligenin, 89.
- Gitalin, 89.
- Gitogenin, 89.
- Gitonin, 89.
- Glaucophyllin, 107.
- Gliadin, 165, 170, 176.
- Globulins, 170, 175, 176.
- Glucase, 186.
- Glucodecose, 44.
- Glucoheptose, 36, 44.
- Glucononose, 36.
- Glucooctose, 36.
- Glucoproteins, 175.
- Glucose, 23, 28, 32, 36, 37, 40, 41, 42, 43, 44, 45, 46, 57, 77.
- Glucosidases, 189.
- Glucosides, 18, 20, 41, 48, 55, 76, 91, 93.
- Glue, 210.
- Glutelins, 175, 176.
- Glutenin, 176.
- Glycerine (see glycerol.)
- Glycerol, 129, 131, 134, 142.
- Glycine, 166, 168, 177.
- Glycinin, 176.
- Glycogen, 59, 61.
- Glycyphyllin, 79.
- Graminin, 59, 62.
- Granulose, 60.
- Grape sugar (see glucose.)
- Guanase, 190.
- Guanine, 160, 162.
- Gulose, 36, 37.
- Gum arabic, 68.
- Gums, 62, 67, 68.
- Gum tragacanth, 69.
- Gun-cotton, 73.
Hæmatin, 110.- Hæmatinic acid imide, 109.
- Hæmatoporphyrin, 110.
- Hæmoglobin, 110.
- Hæmopyrrole, 109.
- Helicin, 81.
- Hemi-celluloses, 63, 71.
- Hemi-terpenes, 147.
- Heptoses, 30.
- Hesperidin, 79.
- Hesperitin, 79, 80.
- Heterotrophic plants, 16.
- Hexosans, 59, 67.
- Hexoses, 22, 28, 30.
- Histidine, 169, 177.
- Histones, 175, 176.
- Honey sugar (see fructose.)
- Hordein, 153, 170, 176.
- Hormones, 92, 239, 240.
- "Hot-drawn oils," 137.
- Humins, 67.
- Hydrastine, 155.
- Hydrazones, 40, 49.
- Hydrocellulose, 73.
- Hydrogen peroxide, 26, 27, 190.
- [Pg 264]Hydrogel, 205.
- Hydrolases, 186, 189.
- Hydroquinone, 77, 79.
- Hydrosol, 205.
- Hydroxy-phenyl ethyl amine, 153.
- Hygrine, 155, 156.
- Hyoscine, 155.
- Hyoscyamine, 156.
- Hypoxanthine, 160.
Idain, 85.- Idose, 36, 37.
- Illuminating gas as a toxin, 247.
- Imbibition, 247.
- Impermeable membranes, 228.
- Indian yellow, 84.
- Indican, 78, 85.
- Indigo, 78, 84.
- Indigotin, 85.
- Indole, 158.
- Indoxyl, 85.
- Inhibitors, 196.
- Intracellular enzymes, 184.
- Inulin, 59.
- Inulinase, 62, 189.
- Invertase, 50, 189, 191.
- Invert sugar, 47, 50.
- Iodine number, 138.
- Ionization phenomena, 226.
- Iridin, 79.
- Irigenin, 79, 80.
- Iron, 3, 5, 11, 110.
- Isochlorophyllin a, 106, 107, 108.
- Isochlorophyllin b, 106, 107, 108.
- Isohæmopyrrole, 138.
- Isoleucine, 168.
- Isomaltose, 51.
- Isomerism, 32.
- Isoprene, 147.
- Isoquercitrin, 84.
- Isoquinoline, 155.
Jalapin, 81.- Japan wax, 129.
- Juglansin, 176.
Ketose, 32.
Lactam, 104.- Lactase, 56.
- Lactic acid ferment, 190.
- Lactone, 104.
- Lactose, 45, 49, 52.
- Laudanosine, 158.
- Laudanum, 158.
- Lactose, 7, 141, 142, 143.
- Lecithoproteins, 175.
- Legumelin, 176.
- Legumin, 170, 176.
- Leucine, 115, 168, 177.
- Leucomaines, 152.
- Leucosin, 176.
- l-galactose, 33.
- l-glucose, 33.
- Levulosans, 59, 62.
- Levulose (see fructose.)
- Lichenin, 62.
- Light, 21, 253, 257.
- Lignocelluloses, 74.
- Lignose, 31.
- Limettin, 82.
- Limonene, 147.
- Linalyl acetate, 148.
- Linseed oil, 133.
- Lipases, 186, 189.
- Lipins (see lipoids.)
- Lipoids, 129, 140.
- Lipoproteins, 175.
- Lupinine, 155.
- Lycopersicin, 102, 122, 144.
- Lysine, 169, 171, 177.
- Lyxose, 35.
Magnesium, 3, 5, 9, 10, 11, 13, 14, 68.- Maltase, 55, 184, 189.
- Maltose, 45, 49, 51, 52.
- Malvidin, 85.
- Malvin, 85.
- Mandelo-nitrile, 87, 88.
- Mandelo-nitrile glucoside, 77, 87.
- Manganese, 4, 13.
- "Manna," 47.
- Mannans, 59, 62, 63, 72.
- Mannite, 47.
- Mannitol, 47.
- Mannitol series, 36.
- [Pg 265]Mannoheptose, 36, 44.
- Mannononose, 47.
- Mannooctose, 36.
- Mannosans (see mannans.)
- Mannose, 32, 36, 37, 41, 44, 45, 47, 57, 72.
- Mannosides, 42.
- Maple sugar (see sucrose.)
- Maysin, 176.
- Melibiose, 49, 52.
- Melizitose, 52.
- Menthol, 148.
- "Mercerizing" cotton, 73.
- Mercury, 247.
- Merosinigrin, 88.
- MaltoseMetallic salts, 13, 224, 227, 247.
- "Metal proteids,", 14.
- Meta-pectin, 70.
- Metaproteins, 175.
- Methylethylmalein imide, 108.
- Methyl glucosides, 42.
- Methyl pentoses, 35.
- Methyl salicylate, 81.
- Middle lamella, 67, 70.
- Millon's reaction, 173.
- Molisch's reaction, 174.
- Monoglycerides, 131.
- Monohydric alcohols, 135.
- Monosaccharides, 31, 35, 45.
- Morin, 119.
- Morphine, 155, 158.
- Morphological adaptations, 251, 252, 255.
- Mucilages, 67, 70.
- Muco-celluloses, 74.
- Muscarine, 152.
- Mustard oils, 88, 148.
- Mustard oil glucosides, 88.
- Mutarotation, 46, 49.
- Myrosin, 77, 88, 149, 189.
- Myrtillidin, 85.
- Myrtillin, 85.
Narceine, 158.- Narcotine, 158.
- "Natural selection,", 254.
- Neurine, 152.
- Nicotine, 155, 156.
- Nitrates, 6.
- Nitrile reaction, 43.
- Nitriles, 43, 44.
- Nitrogen, 3, 5, 6, 151, 164.
- Non-drying oils, 132.
- Non-essential elements, 4.
- Non-reducing sugars, 39, 49.
- Nonoses, 31.
- Normal celluloses, 72.
- Nuclease, 189.
- Nucleoproteins, 162, 175.
- Nutrients, 1.
Octoses, 31.- Œnidin, 85, 116.
- Œnin, 85.
- Oils, 129.
- Oil of bergamot, 148.
- Oil of bitter almonds, 86, 148.
- Oil of cassia, 148.
- Oil of cinnamon, 148.
- Oil of garlic, 148.
- Oil of lavender, 148.
- Oil of mustard, 148.
- Olive oil, 130.
- Opium, 158.
- Organic acids, 18, 124, 248.
- Organized ferments, 183.
- Ornamental pigments, 102, 123.
- Ornithine, 169.
- Oryzenin, 176.
- Osazones, 40, 41, 49.
- Osmotic pressure, 213, 228.
- Osones, 41.
- Oxidases, 186, 190, 193.
- Oxime, 44.
- Oxycellulose, 73.
- Oxycumarin glucosides, 81.
- Oxygenated oils, 147.
- Oxyhydroquinone, 95.
- Oxyproline, 169.
Pæonidin, 85.- Pæonin, 85.
- Palm oil, 133.
- Papain, 189.
- Papaverine, 155, 158.
- Para-dextran, 62.
- [Pg 266]Para-isodextran, 62.
- Paralyzers, 196.
- Para-pectin, 70.
- Parasites, 16.
- Peanut oil, 133.
- Pectase, 71.
- Pectinase, 189.
- Pectins, 20, 31, 67, 70.
- Pecto-celluloses, 74.
- Pectose, 31, 70.
- Pelargonidin, 85, 116.
- Pelargonin, 85.
- Pentosans, 31, 67, 68, 72.
- Pentoses, 30, 162.
- Pepsin, 167.
- Peptids, 166, 167, 176.
- Peptones, 176.
- Permeable membranes, 228.
- Peroxidases, 190.
- Persimmons, 100.
- Persuelose, 36.
- Phæophytin, 107, 108.
- Phaselin, 176.
- Phaseolin, 176.
- Phenol, 95.
- Phenol glucosides, 79.
- Phenyl alanine, 168, 177.
- Phenyl hydrazine, 40.
- Phlein, 62.
- Phloretin, 79.
- Phloridzin, 79.
- Phloroglucinol, 95.
- Phosphates, 7.
- Phosphatides, 141, 143.
- Phosphoproteins, 175.
- Phosphorus, 3, 5, 7.
- Photochemical action of light, 253, 257.
- Photolysis, 26.
- Photosynthesis, 7, 8, 18, 21, 22, 24, 254.
- Phycoerythrin, 102, 115.
- Phycophæin, 102, 115.
- Phyllins, 106, 107.
- Phyllophyllin, 107.
- Phyllopyrrole, 109.
- Physiological adaptations, 252, 257.
- Physiological use, 19.
- Phytase, 189.
- Phytochlorin, 108.
- Phytorhodin, 108.
- Pigment glucosides, 82.
- Pigments, 18, 102, 224, 254.
- Pinene, 147.
- Piperidine, 154.
- Piperine, 155.
- Plant amines, 151, 152, 163.
- Plant food, 1.
- Plant nucleic acids, 162.
- Polybasic acids, 124.
- Polyhydric alcohols, 31.
- Polypeptides, 167.
- Polysaccharides, 59.
- Polyterpenes, 147.
- Poppy wax, 140.
- Populin, 80.
- Porphyrins, 108.
- Potassium, 3, 5, 8, 10, 13, 14.
- Primary amines, 152.
- Proenzymes, 198.
- Proinulase, 199.
- Proinvertase, 199.
- Prolamins, 175, 176.
- Proline, 169, 177.
- Prolipase, 199.
- Prooxidase, 199.
- Protamins, 175, 176.
- Proteans, 175.
- Proteases, 186, 189, 192.
- Protective colloids, 209.
- Proteins, 7, 18, 20, 151, 162, 163, 164, 224.
- Proteoses, 175.
- Protoplasm, 17, 26, 221.
- Prulaurasin, 87.
- Prunase, 87.
- Prunasin, 87.
- Ptomaines, 152.
- Purine, 159.
- Purine bases, 151, 159, 162.
- Purpurin, 83.
- Putrescine, 152.
- Pyrimidine, 161.
- Pyrimidine bases, 161, 162.
- Pyrocatechol, 95.
- Pyrogallol, 95.
- [Pg 267]Pyrogallol tannins, 97.
- Pyroxylin, 73.
- Pyrrophyllin, 107.
- Pyrridine, 154.
- Pyrrolidine, 154.
Quaternary amines, 152.- Quercetin, 84, 118.
- Quercitrin, 84.
- Quinine, 155, 157.
- Quinoline, 155, 158.
Radiant energy, 19.- Raffinose, 45, 52, 53.
- Rape-seed oil, 130.
- Reducing sugars, 39, 49.
- Reductases, 186, 190.
- Reserve food, 21.
- Resenes, 149.
- Resins, 18, 146, 149.
- Resorcinol, 95.
- Respiration, 18, 121, 235, 236.
- Rhamnase, 77, 189.
- Rhamnetin, 84.
- Rhamnose, 35, 52, 77, 79.
- Rhodeose, 35, 81.
- Rhodophyllin, 107.
- Ribose, 35.
- Ricin, 176.
- Rubiadin, 83.
- Rubiphyllin, 107.
Saccharide, 31.- Salicin, 80.
- Saligenin, 80.
- Salinigrin, 81.
- Salts, 224, 227, 237.
- Sambunigrin, 87.
- Sapogenins, 90.
- Saponification, 134.
- Saponification value, 138.
- Saponins, 90.
- Sapotoxins, 90.
- Saprophytes, 16.
- "Saturated" acids, 132.
- Scopolin, 82.
- Secalin, 63.
- Secondary amines, 152.
- Secretions, 20.
- Sedoheptose, 36.
- Semipermeable membranes, 228.
- Sensitizers, 27.
- Serine, 168.
- Silicates, 12.
- Silicon, 4, 12.
- Silver, 247.
- Simple proteins, 165, 174, 175.
- Sinalbin, 89.
- Sinalbin mustard oil, 89.
- Sinapin acid sulfate, 89.
- Sinigrin, 88.
- Sinistrin, 62.
- Sitosterol, 136.
- Skimmetin, 81, 82.
- Skimmin, 81.
- Soaps, 81134, 208.
- Sodium, 4, 9, 12, 13, 14.
- Sodium stearate, 133.
- Sol, 205.
- "Soluble starch,", 60.
- Sorbitol, 48.
- Sorbose, 16, 38, 45, 48.
- Specific rotatory power, 38, 39.
- Skimmetin, 129, 133.
- Stachydrine, 135.
- Stachyose, 54.
- Starch, 8, 22, 28, 30, 31, 45, 59, 64.
- "Starch paste," 60.
- Stearin, 131, 134.
- Stereo-isomerism, 32.
- Stigmasterol, 136.
- Structural adaptations, 255.
- Structural isomerism, 32.
- Strychnine, 155, 157.
- Substrate, 186.
- Sucrase (see invertase.)
- Sucrases, 186.
- Sucrose, 28,49, 64.
- Sugars, 8, 18, 22, 28, 30, 31.
- Sulfur, 3, 5, 11, 148.
- [Pg 268]Sulfuretted oils, 147, 148.
- Sulfur test, 186.
- Sunflower-seed oil, 130.
- Surface boundary phenomena, 231.
- Surface energy, 231.
- Surface tension, 231.
- "Survival of the fittest," 254.
- Suspensoids, 206, 214.
- Suspensions, 206.
- Synergic foods, 2, 20.
- Suspensions, 18.
Tagatose, 36, 38, 57.- Talose, 36, 38, 42, 57.
- Tannins, 18, 94, 97, 99, 100, 127, 208, 224.
- Tannon group, 96.
- Terpenes, 147.
- Tertiary amines, 152.
- Tetrapeptides, 167.
- Tetrasaccharides, 54.
- Tetrose, 30, 35.
- Theobromine, 160.
- Theophylline, 160.
- Thioglucose, 88.
- Threose, 35.
- Thymine, 161, 162.
- Thymol, 148.
- Toxins, 13, 239, 240, 245.
- Translocation diastase, 191.
- Trehalase, 51.
- Trehalose, 49, 50.
- Triglycerides, 131.
- Trimethyl amine, 152.
- Trimethyl glycocoll, 143.
- Triose, 30, 35.
- Trioxymethylene, 22, 23.
- Tripeptides, 167.
- Trisaccharides, 31, 52.
- Triticin, 59, 62.
- Tryptophane, 169, 171, 177.
- Tuberin, 176.
- Turanose, 49, 53.
- Tyndall phenomena, 212.
- Tyrosine, 115, 168, 177.
Unavailable plant food, 4.- Ultrafilter, 215.
- Ultramicroscope, 203, 204, 205, 211.
- Unorganized ferments, 183.
- "Unsaturated" acids, 132, 138.
- Uracil, 161, 162.
- Urease, 190.
Valine, 168.- Vanillin, 80, 148.
- Vegetable bases, 18, 151.
- Vicianin, 88.
- Vicilin, 176.
- Vignin, 176.
- Vitamines, 239, 240, 242.
- Volatile oils, 20, 147.
Water, 3, 21, 22, 23, 224.- Water-soluble B, 243.
- Waxes, 18, 129, 140.
- Weathering, 4.
- Wood gum, 68.
- Wool fat, 129.
- Wound gum, 68, 69.
Xanthine, 160.- Xanthone, 82, 83, 102.
- Xanthophyll, 112, 113, 121.
- Xanthopurpurin, 83.
- Xanthorhamnin, 52, 84.
- Xylan, 69.
- Xylose, 35, 68, 69.
- Xylosides, 56.
Yeast, 61.
Zein, 165, 170, 176, 177.- Zinc, 13.
- Zymase, 51, 56, 190, 192.
- Zymogens, 198.
